

信息中心


2022年12月6日,国际学术期刊Nature刊发了上海交通大学变革性分子前沿科学中心申涛副教授、福州大学叶克印教授及康奈尔大学Tristan Lambert教授等人合作的科研成果“Electrophotocatalytic Oxygenation of Multiple Adjacent C–H Bonds”。申涛副教授为第一作者,叶克印教授及Tristan Lambert教授为共同通讯作者。

与发展成熟的烯基双官能团化相比,烷基的官能团化往往局限于单一位点的单官能团化,烷基连续相邻碳氢键的直接双官能团化及多官能团化则非常挑战。这主要由于:1. 单官能团化后对邻位碳氢键活性的钝化作用;2. 连续多官能团化中过度氧化降解导致合成效率低;3. 多官能团化的区域选择性,化学选择性及立体选择性调控。
氧原子及含氧官能团广泛存在于在天然产物,药物,材料中,对人类的衣食住行产生了深远影响。在自然界中,生物体通过酶催化的方法以高选择性的建立多个碳氧键,但与生物合成相比,通过化学合成同时实现连续惰性碳氢键的氧化往往面临邻位碳氢键活性钝化及过度氧化的挑战性问题,因此亟需一种足够强大但又足够温和的反应策略。
基于此,上海交通大学申涛副教授、福州大学叶克印教授与康奈尔大学Tristan Lambert教授报告了通过光电共催化策略实现两个或三个连续的C-H键的选择性氧化,使简单的烷基苯或三氟乙酰胺转化为相应的双氧化及三氧化产物。值得一提的是,该过程可以通过酸添加剂的改变选择性地合成二氧化或三氧化产物。
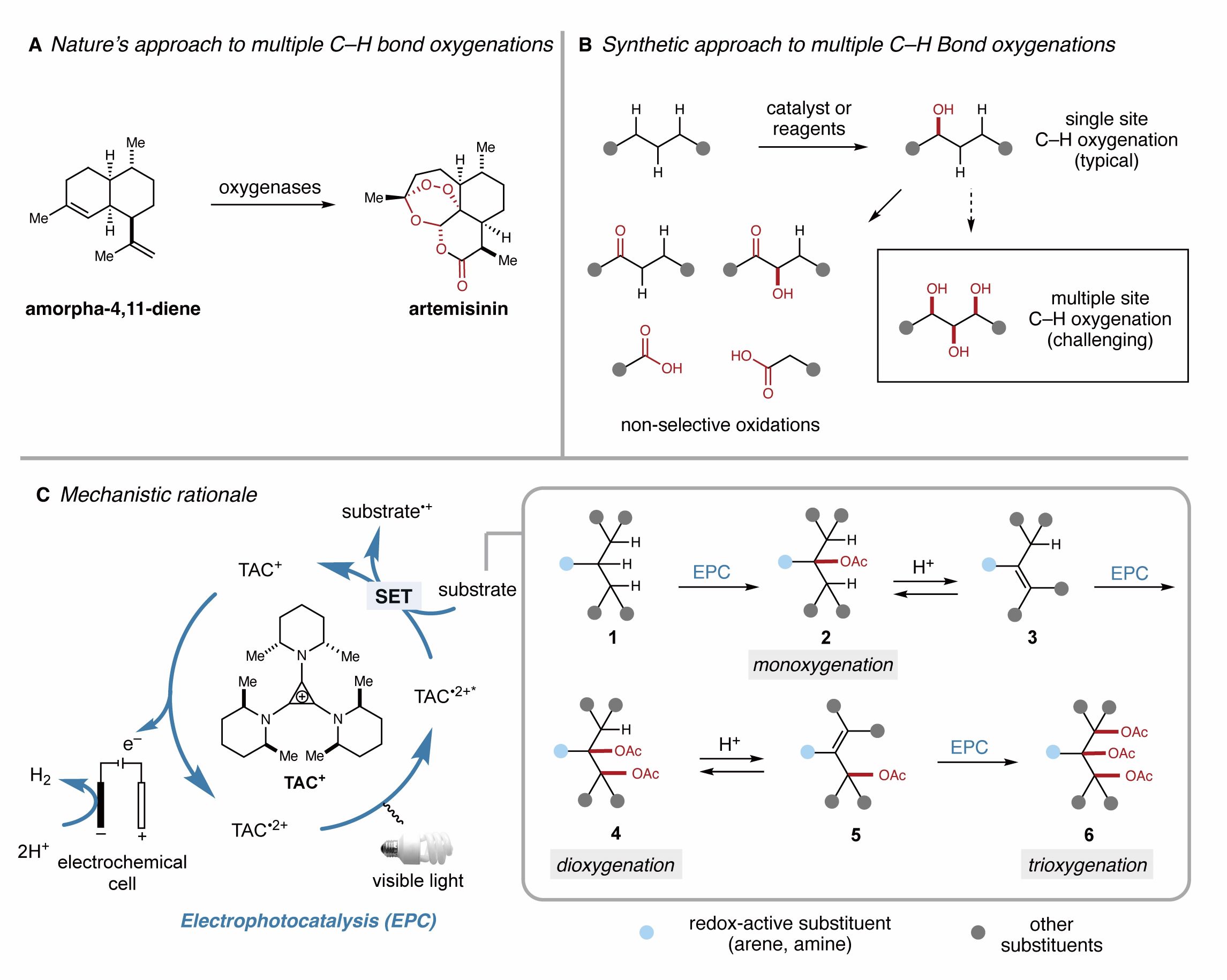
图1. 连续相邻多个碳氢键氧化的挑战和策略
在申涛副教授及Tristan Lambert教授之前合作的光电共催化烷基邻位碳氢键双胺化工作(Shen, T., Lambert, T. H. Electrophotocatalytic diamination of vicinal C–H bonds. Science 2021, 371, 620-626.)基础上, 作者设想可以利用连续循环的氧化-消除途径在烷基双官能团化基础上进一步实现烷基的连续碳氢键的三官能团化(三氧化)。而其中的关键是酸添加剂和亲核试剂的选择,使得反应中间体发生可逆缓慢的消除烯化,以低浓度进行双氧化反应进而避免烯烃中间体的聚合和降解(图1C)。
在筛选了大量的酸和含氧亲核试剂后,作者意识到亲核试剂浓度的重要性,最终作者选择将乙酸、醋酐(Ac2O)及二氯甲烷作为混合溶剂进行电解。强酸的选择对于消除烯化过程非常重要,对于三级烷基苯,使用较弱的三氟乙酸即可完成苄位消除烯化过程,进而顺利发生双氧化反应;而对于二级烷基苯,其消除烯化更难,需采用更强的三氟甲磺酸作为添加剂反应才得以发生(见SI)。随后作者采用光电共催化的方法,使用TAC+ClO4-(8 mol%)作为催化剂进行简单烷基苯的双氧反应(图2)。该转化有着很好的官能团兼容性,例如容易氧化的卤素、乙酰氧基、酯基、酰亚胺、醇、羧酸等可以顺利得到产物,甚至非保护的三级胺亦可耐受。产物经过简单一步水解后即可得到用途广泛的邻二醇类化合物,简洁高效。一些活泼易氧化的杂环,如噻吩,呋喃,噻唑,喹啉,吖啶等也可以兼容当前光电催化的条件。其他的酸作为溶剂反应也可以顺利进行。通常情况下,产物构型以anti为主。

图2. 连续邻位碳氢键双氧化
由于较弱的三氟乙酸即可实现三级烷基苯的双氧化反应,作者设想,如果使用更强的酸,能否发生连续的E1型消除氧化,进而实现第三个碳氢键的氧化。通过对强酸的筛选,作者发现当使用三氟甲磺酸HOTf时,确实可以顺利得到简单烷烃的三氧化产物。值得一提的是,这是第一例发生连续的C-H三氧化反应的报告(图3)。一系列简单取代的三级烷基苯都可以顺利发生该转化,而且可以实现克级快速制备(53, 1.86 g), 产物经过简单水解后即可生成用途广泛的邻三醇。由于位阻影响,一些产物中苄位被醇羟基取代。一些易氧化降解的杂化官能团如呋喃,噻吩,喹啉,吖啶等均可兼容。该方法为快速制备邻三醇类化合物提供了新的思路。

图3. 连续邻位碳氢键三氧化
随后作者设想,第一次的氧化启动是非常关键的,需要有活泼官能团作为“定位基团”,因此可以将反应体系进一步拓展为“基于活性位点启动的烷基多官能团化”。除芳基外,其他的活泼杂原子如N, O, S的邻位等理论上都有发生该类反应的可能。经过大量尝试,作者发现三氟乙酰基保护的胺类化合物如哌啶,吡咯烷,二级胺等也可以进行多个邻位碳氢键氧化反应(图4A)。有趣的是,哌啶类底物可以顺利生成三氧化产物,而其他类型的胺类则以双氧化产物为主。作者小心谨慎,用半年之久培养出三个化合物的单晶(79, 84, 86), 最终确定反应产物的构型。大多数产物以以良好的非对映选择性生成。这是一个相当令人惊讶的反应,体现了电化学独特的反应性,为生物活性分子氮杂糖的直接一步构建提供了新的策略。
药物活性分子的后期修饰为优化药物性质及发现新的药物提供了简洁快速的方法,然而药物的后期修饰往往依赖当前所发展的官能团转化方法。烷基在药物分子中无处不在,虽然烯烃双官能团化方法发展成熟,但无法用于无烯基但有烷基的药物分子,该策略提供了一种对该类分子进行直接双官能团化/多官能团化的修饰方法,成为烯基双官能团化的重要补充。作者将这类新颖的双氧化及三氧化反应应用于一系列更复杂的药物活性分子的衍生化中(图4B)。该策略顺利实现了香精和香料剂celestolide的简单双氧化,克级制备产率为69%(2.5g,10mmol)。此外,一些药物分子例如受体激动剂、氟比洛芬等,都有很好的产率。值得一提的是,该反应甚至可以实现一步的8电子,10电子,12电子氧化,直接实现底物的四官能团化,五官能团化甚至六官能团化,充分体现了该体系强大的反应性。
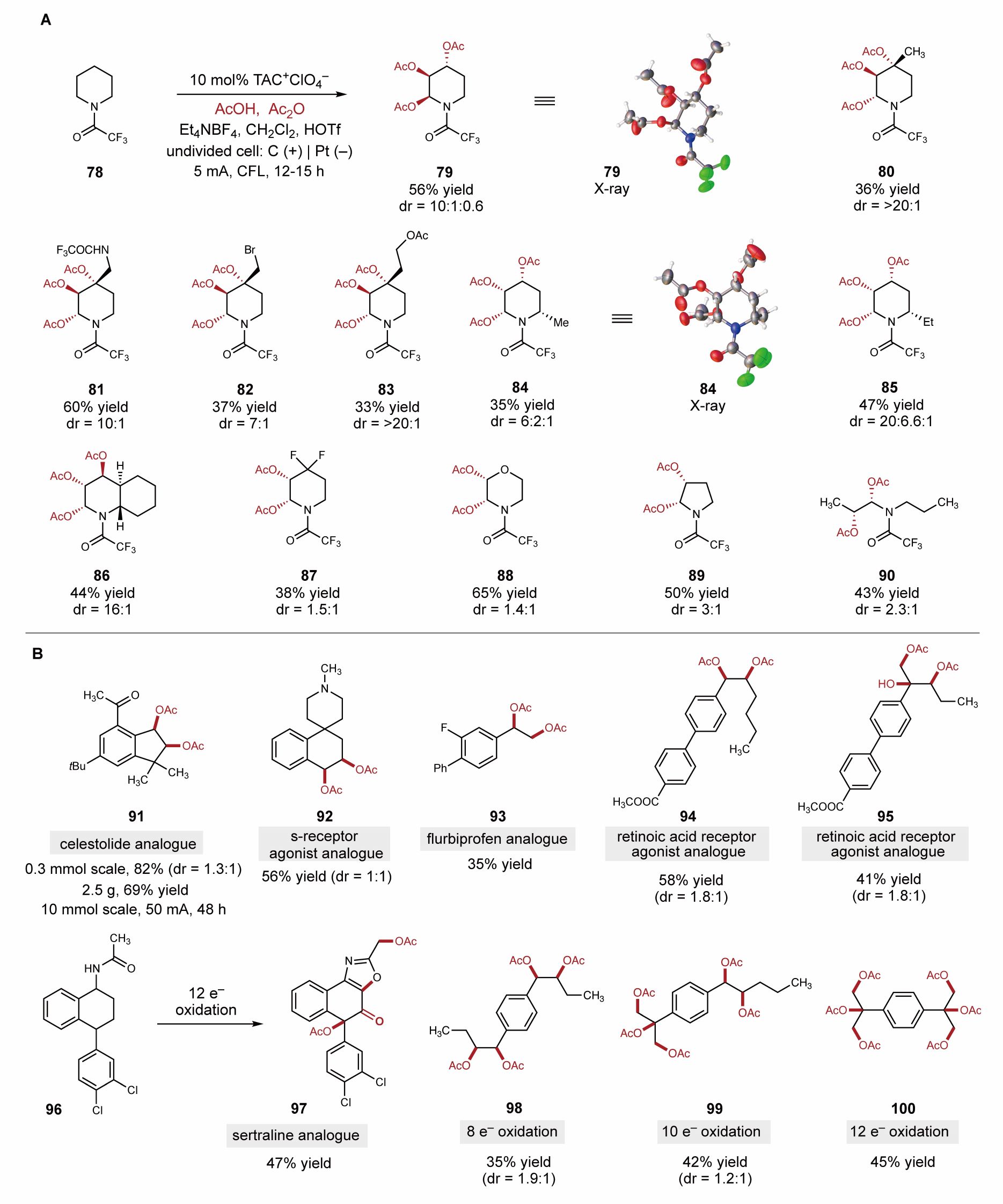
图4. 连续邻位碳氢键氧化的应用
为了验证该策略在实际合成中的应用,作者初步尝试了某些已知药物分子的合成(图5)。例如可以3步44%的产率合成抗菌药物京纳康唑的中间体,而传统方法需要8步,产率仅为8%;一步46%产率合成氮杂糖衍生物105,而传统方法则需要5步,产率36%;最后作者以3步42%产率合成了vanilloid受体配体的中间体,以往方法则需要5步,产率仅为11%。因此,显而易见在一次操作中实现多个连续的C-H氧化反应可以大大简化复杂分子的合成路线并提高产率。

图5. 连续邻位碳氢键氧化在简化药物分子合成中的应用
总结:该工作首次实现了邻位连续碳氢键的多氧化反应,发展了新颖的光电共催化的连续消除-氧化策略,开创了邻位烷基碳氢键多官能团化的先河,为快速增加分子复杂度和药物分子后期修饰及简洁合成提供的新的思路。
【作者简介】

申涛,2012年本科毕业于中国农业大学理科基地班,2017年北京大学获博士学位(导师:焦宁教授),2018年至2022年于美国CornellUniversity进行博士后研究(合作导师:Tristan Lambert教授),现任上海交通大学变革性分子前沿科学中心课题组长、长聘教轨副教授、博士生导师。主要研究方向:惰性键的选择性活化与重组,绿色合成化学。近年来发表论文20余篇,其中以第一作者在Nature (1篇), Science (1篇),J. Am. Chem. Soc. (2篇),Angew. Chem. Int. Ed.(1篇) 等重要学术期刊发表研究论文10余篇。个人网页地址:https://fsctm.sjtu.edu.cn/info/1011/1553.htm

叶克印,福州大学化学学院闽江学者特聘教授,福建省百人计划专家。研究方向包括电化学合成与催化、不对称催化、药物化学。学术期刊 Chinese Chemical Letters,Green Synthesis & Catalysis 青年编委。

Tristan Lambert,康奈尔大学教授,化学系副主任。研究主要集中于催化剂设计,发展了电光催化、催化羰基烯烃复分解和手性芳香离子催化的新型方法。




