

科学研究


近日,上海交通大学变革性分子前沿科学中心商明团队在脱氧官能化领域取得重要进展,发展了一种“极性-自由基转换”新范式,实现了叔醇的直接脱羟基烷基化反应,为构建具有重要应用价值的全碳季碳中心提供了一条通用、高效的新路径。该研究成果以"Quaternary Carbon Centers via Electrochemical Direct Dehydroxylative Alkylation"为题发表在Angew. Chem. Int. Ed.上。
全碳季碳中心,即连接四个碳原子的碳中心,作为天然产物与药物分子的核心结构单元,因其独特的空间位阻效应与结构刚性,被证实能够显著提升药物分子的代谢稳定性及靶标结合力。因此,全碳季碳中心的构建始终是有机合成领域的研究热点。叔醇由于其易得性、化学稳定性以及丰富的天然存在形式,被视为构建季碳中心的理想前体。然而,C–OH键的高键能和羟基较差的离去能力限制了其在脱氧构建C(sp³)–C(sp³)键反应中的应用。
为了克服这一难点,脱氧活化前体策略被提出。自1985年Barton首先发展了2-巯基吡啶衍生的脱氧前体后,多种自由基脱氧前体在近20年得到了蓬勃的发展。通过对前体骨架的改变,其化学活性和自由基引发方式更具多样性,增加了该类反应的应用场景。尽管该策略在三级醇脱氧官能化领域取得较大成功,其在步骤经济性方面的不足一直是化学家想要解决的一个问题。因此,脱氧反应的研究焦点逐渐转向三级醇的直接或原位活化策略,通过避免自由基前体的合成与分离,提高反应的效率和经济性。但是由于C–O键本身的惰性,目前原位或直接活化策略仍然存在原子经济性低,操作繁琐,条件不够温和等问题,有较大探索与提升的空间。
为了解决这一难题,商明团队独辟蹊径,提出了一个"极性到自由基转化"的活化新范式。在路易斯酸存在下,三级醇发生 C–OH键的异裂,得到高活性的碳正离子物种。通过亲核性阴离子与碳正离子结合,对其进行稳定,减少消除反应的发生。随后该加合物在体系中经过单电子转移过程,离去该亲核性离子,得到碳自由基中间体。这一策略如同搭建了一座桥梁,让易于生成的碳正离子能够被转化为更具偶联活性的碳自由基,从而为攻克三级醇直接脱氧官能团化,特别是构筑拥挤的季碳中心,开辟了一条富有潜力的新途径。
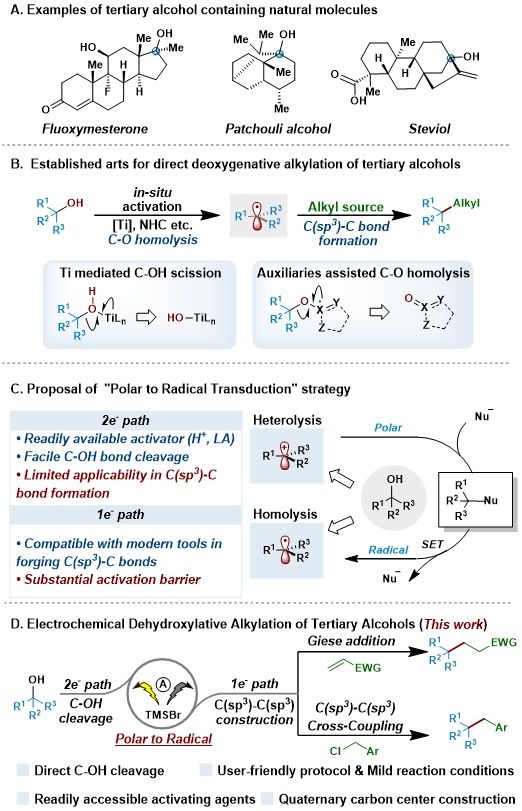
图1. 研究背景
作者通过Giese型自由基加成反应来验证其设计思路,以1,1-二甲基-3-苯基丙醇(1)和丙烯酸甲酯(2)为模板底物,对反应条件进行了系统优化(图2)。最终确定以TMSBr为活化试剂,Ag₂O为添加剂,在乙腈溶剂中,采用Fe阳极,泡沫Ni阴极,在恒电流(10 mA)条件下,能够以78%的分离收率获得目标烷基化产物。控制实验证明了TMSBr、电流和Ag₂O对于反应成功至关重要。
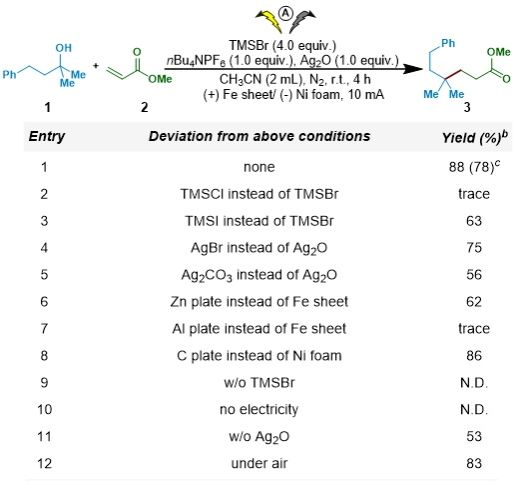
图2. 条件筛选
在最优条件下,作者对该策略的底物普适性进行了探索,该策略展现了优异的普适性和官能团耐受性:无论是带有不同芳基和烷基取代基的开链叔醇,还是不同大小的环状叔醇(包括杂环底物),都能顺利参与反应。在二醇底物中,能够选择性活化叔羟基,而伯、仲羟基不受影响。从药物分子和天然产物衍生得到的结构复杂的叔醇以及缺电子烯烃,也能高效转化为相应的脱羟基烷基化产物,凸显了该方法在药物研发和天然产物合成中进行后期官能团化的巨大潜力。除了丙烯酸酯类迈克尔受体,丙烯酰胺、丙烯腈、环烯酮等其他类型缺电子烯烃也能良好兼容。
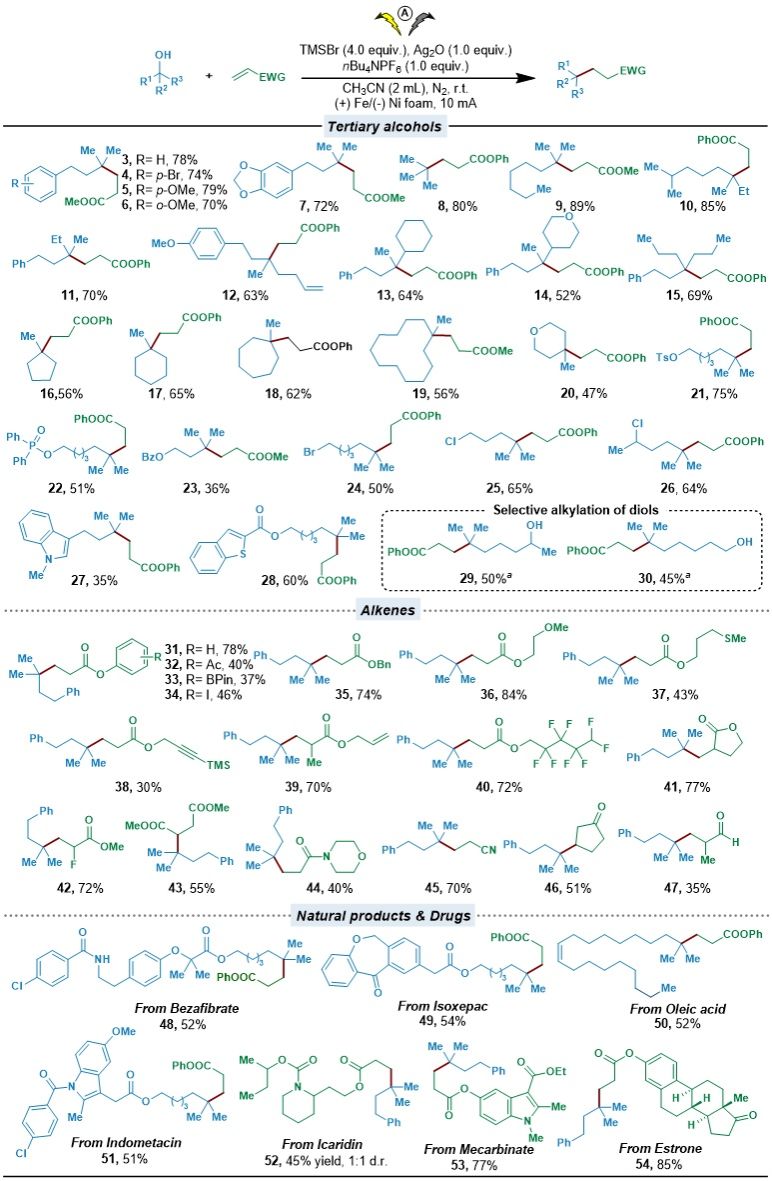
图3. Giese型自由基加成反应底物拓展
此外,作者还将该策略拓展至与苄氯的镍催化脱羟基交叉偶联反应(图4)。
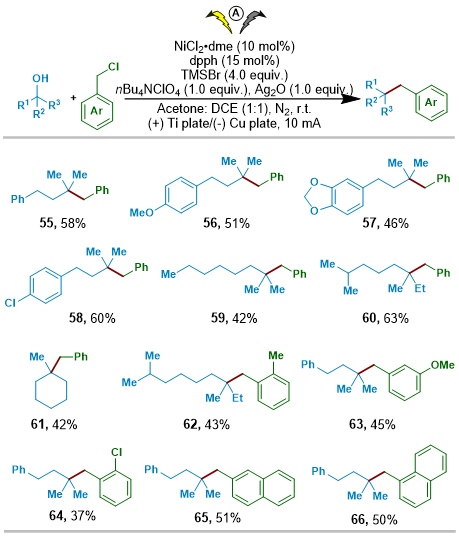
图4. Ni催化偶联反应底物拓展
为了展示该方法在合成中的应用潜力,作者展示了规模放大合成(图5A),且可降低Ag₂O用量。同时,作者选取了两个已报道的目标化合物,运用电还原直接脱氧烷基化策略为关键步骤,以更简洁的步骤和更廉价的起始原料对其进行合成,进一步展示该方法在合成中的高效性(图5B)。为了深入理解反应机理,作者进行了一系列机理实验(图5C-F):自由基捕获实验,碳正离子探究实验,活性中间体探究实验以及循环伏安实验,验证了碳正离子到碳自由基转化的过程。
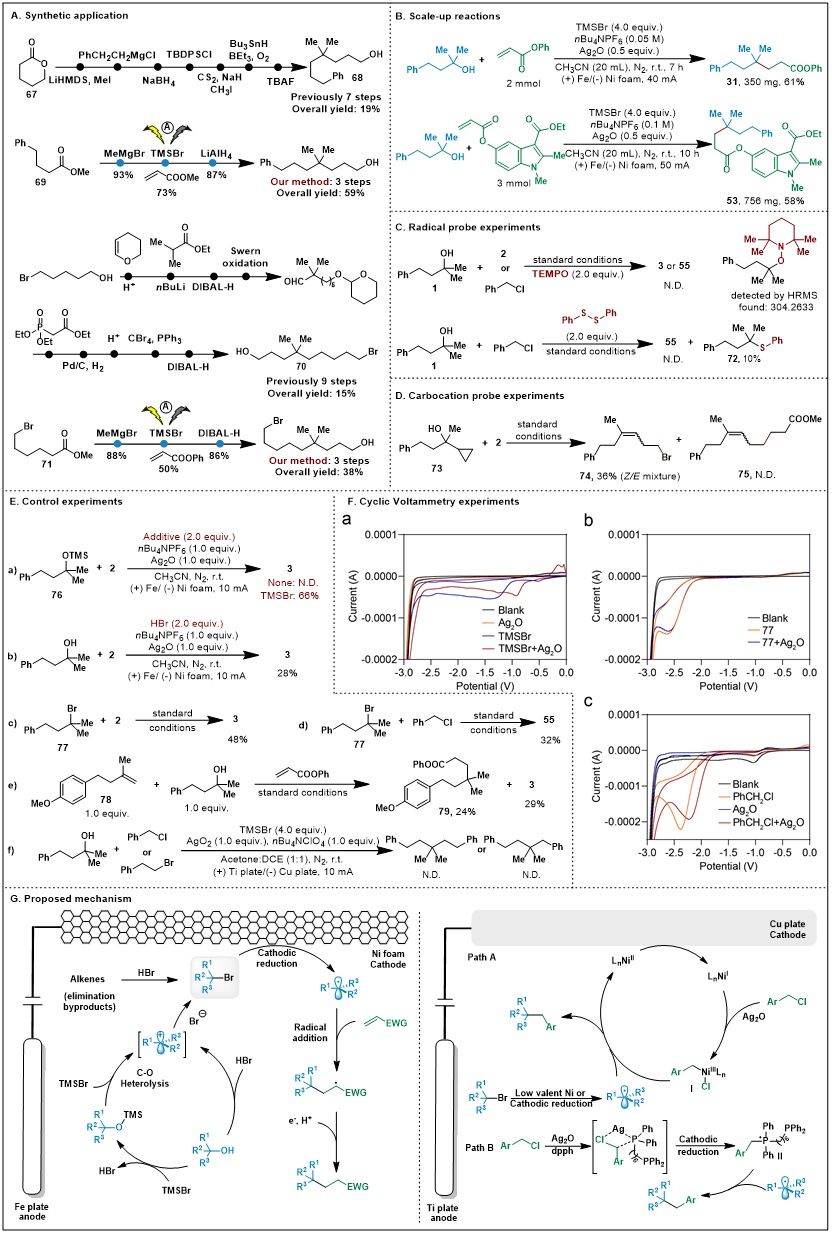
图5. 合成应用与机理研究
综上,这项研究发展了一种电化学三级醇脱羟烷基化反应,通过"极性到自由基转化"策略,成功实现了叔醇与缺电子烯烃及苄氯的偶联,高效构建了全碳季碳中心。该方法条件温和、试剂简单、操作简便,具有广阔的底物适用范围和出色的官能团耐受性,适用于复杂生物活性分子的后期修饰。该工作为惰性C–OH 键的定向转化提供了新思路,也展示了融合不同反应模式以解决合成难题的巨大潜力。




