

科学研究


近日,上海交通大学变革性分子前沿科学中心叶天南团队取得重要进展,相关研究成果以“Phase Engineering of Mg-Pt Intermetallic Hydrogenation Catalysts for CO Poisoning Resistance”为题发表于期刊Angew. Chem. Int. Ed.上。该工作的第一作者为上海交通大学博士生陆小军、李自闯、上海科技大学张青研究员,通讯作者为上海交通大学变革性分子前沿科学中心叶天南副教授。
1. 全文速览
针对镁基单原子金属间化合物(SAIMCs)晶相结构单一,难以可控调节活性位点电子结构问题,以及由此导致的材料结构与抗CO中毒性能之间构-效关系难以建立的困境,上海交通大学叶天南团队通过控制Mg基底表面Pt纳米粒子的结晶度,有效调节了Mg与Pt之间的反应性金属-载体相互作用,从而实现了Mg29Pt4和Mg3Pt两种不同晶体结构SAIMCs材料的可控制备。在两种Mg-Pt SAIMCs材料中,Pt位点均以单原子形式有序分布于对应的晶体结构中,但其电子结构却存在显著差异,导致二者具有不同的抗CO中毒性能。在含有0.2 vol% CO的喹啉加氢体系中,Mg29Pt4的催化活性较Mg3Pt催化剂提升了21倍。同时,相较于纯H2体系,Mg29Pt4催化剂的抗CO加氢活性保留率高达88%,表现出优异的抗CO中毒性能。机理研究表明,Mg29Pt4催化剂优异的抗CO毒化性能,主要源于其Pt位点具有较高的电子密度和较低的d带中心。这一独特的电子结构能双向抑制CO的吸附过程:既抑制了CO的5σ轨道电子向Pt的5d轨道转移,也抑制了Pt的5d电子反向填充至CO的2π*反键轨道,从而显著弱化了CO在Pt位点上的吸附强度,最终提升了催化剂在含CO氢气中的加氢性能。此外,该催化剂在多种有机官能团(如醛基、硝基和炔烃)的加氢反应中均展现出良好的抗CO性能,表明其具有广泛的底物适用性。
2. 背景介绍
铂族金属(PGMs)催化剂在工业加氢过程中具有关键作用,但其活性中心(如Pt)对氢气中痕量的CO极为敏感,容易因CO的强吸附而导致催化剂失活。为避免这一问题,工业上需对氢气进行高能耗的深度纯化,这显著降低了氢能源的经济性。因此,开发具有抗CO中毒能力的PGMs催化剂,兼具重要的科学意义与经济价值。
如图1a所示,Pt基催化剂的CO中毒过程涉及两步电子转移:(1)CO的5σ轨道电子向Pt的5d空轨道转移;(2)Pt的5d电子反向填充至CO的2π*反键轨道。二者共同作用,导致CO在Pt位点上发生强吸附。已有研究表明,单原子催化剂(SACs)在抗CO中毒中发挥着重要作用。从几何结构角度,CO分子通过线性模式吸附在单一金属原子上,这种低配位环境能显著削弱CO的吸附强度。从电子结构角度,缺电子的Pt位点可有效抑制Pt 5d电子向CO 2π*反键轨道填充,从而弱化CO吸附。然而,Pt单原子的缺电子特性也促进了Pt 5d空轨道与CO 5σ轨道之间的杂化,导致CO 5σ电子更加有利地填充至Pt 5d空轨道中(如图1b)。基于对CO中毒机制的深入分析,作者认为富电子态的Pt单原子位点可能更具优势。如图1c所示,富电子态的Pt单原子位点能有效抑制CO 5σ轨道电子向Pt 5d空轨道填充;同时,Pt 5d轨道电子填充可降低Pt位点的d带中心,进一步抑制Pt 5d电子向CO 2π*反键轨道填充,从而弱化CO的吸附。然而,构筑带负电的金属单原子活性中心仍面临巨大挑战。这主要是因为大多数SACs中的金属原子通常与高电负性的p区元素(如N 、O等)配位,使得金属单原子中心往往带正电性。
金属间化合物因其丰富的化学组成、可调的几何与电子结构,在催化领域展现出巨大潜力。在作者先前的工作中(Nat. Catal. 2025, 8, 536-547),成功开发了一系列富镁型Mg24TM4(TM = Pd,Rh,Pt或Ir)单原子金属间化合物(SAIMCs)。在该结构中,过渡金属原子在几何和电子维度上均被Mg原子完全隔离,形成高度有序的单原子位点。同时,由于Mg的电负性较低,电子会转移至TM位点,使其呈现富电子特性。这种独特的结构为解决CO中毒问题提供了新思路。不过,该系列Mg24TM4材料具有固定的晶体结构,难以对其金属活性中心的电子结构进行可控调节。因此,通过晶相工程策略调控TM位点的电子性质,进而建立材料电子结构与抗CO中毒性能之间的构效关系,具有重要的理论意义。
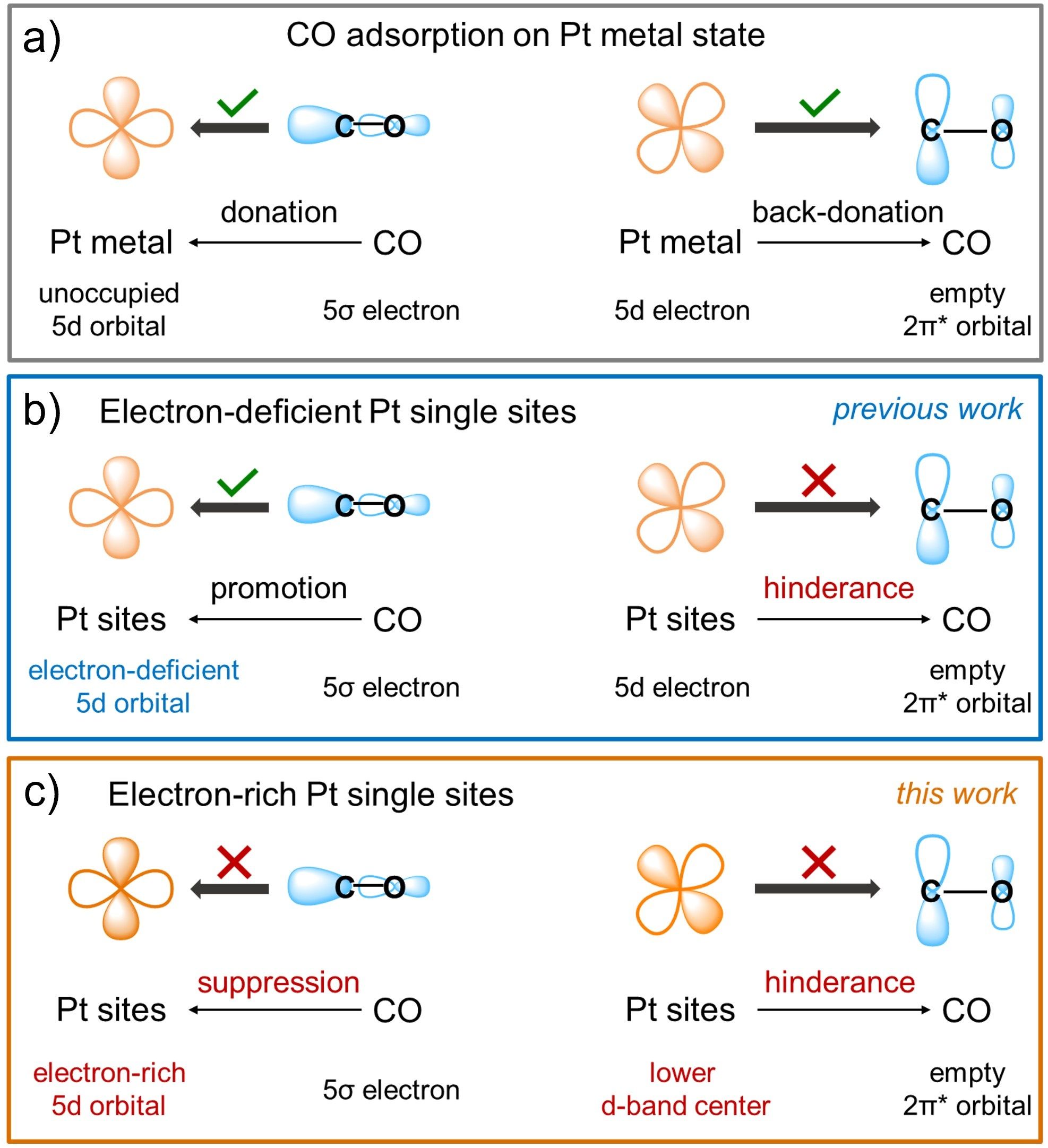
图1. 不同电子结构的Pt位点对CO的响应机制。
3. 本文亮点
(1)通过控制Mg基底表面Pt纳米粒子的结晶度,有效调节了Mg与Pt之间的反应性金属-载体相互作用,从而实现了Mg29Pt4和Mg3Pt两种不同晶体结构单原子金属间化合物材料的可控制备。
(2)在Mg29Pt4和Mg3Pt中,Pt位点均以单原子形式有序分布于对应的晶体结构中,但其电子结构却存在显著差异。其中,富镁型Mg29Pt4中的Pt位点具有更高的电子密度。
(3)在含有0.2 vol% CO的喹啉加氢体系中,Mg29Pt4的催化活性较Mg3Pt催化剂提升了21倍。同时,相较于纯H2体系,Mg29Pt4催化剂的抗CO加氢活性保留率高达88%,表现出优异的抗CO中毒性能。此外,Mg29Pt4催化剂可成功应用于多种有机官能团(如醛基、硝基以及炔烃基团)的抗CO加氢,展现出广泛的底物适用性。
(4)Mg29Pt4催化剂优异的抗CO毒化性能主要源于其Pt位点具有较高的电子密度和较低的d带中心。该电子结构有效抑制了CO 5σ轨道电子向Pt 5d轨道的转移,同时减弱了Pt 5d电子向CO 2π*反键轨道填充,从而显著降低CO在Pt位点上的吸附强度,最终提升Mg29Pt4的抗CO中毒性能。
4. 图文解析
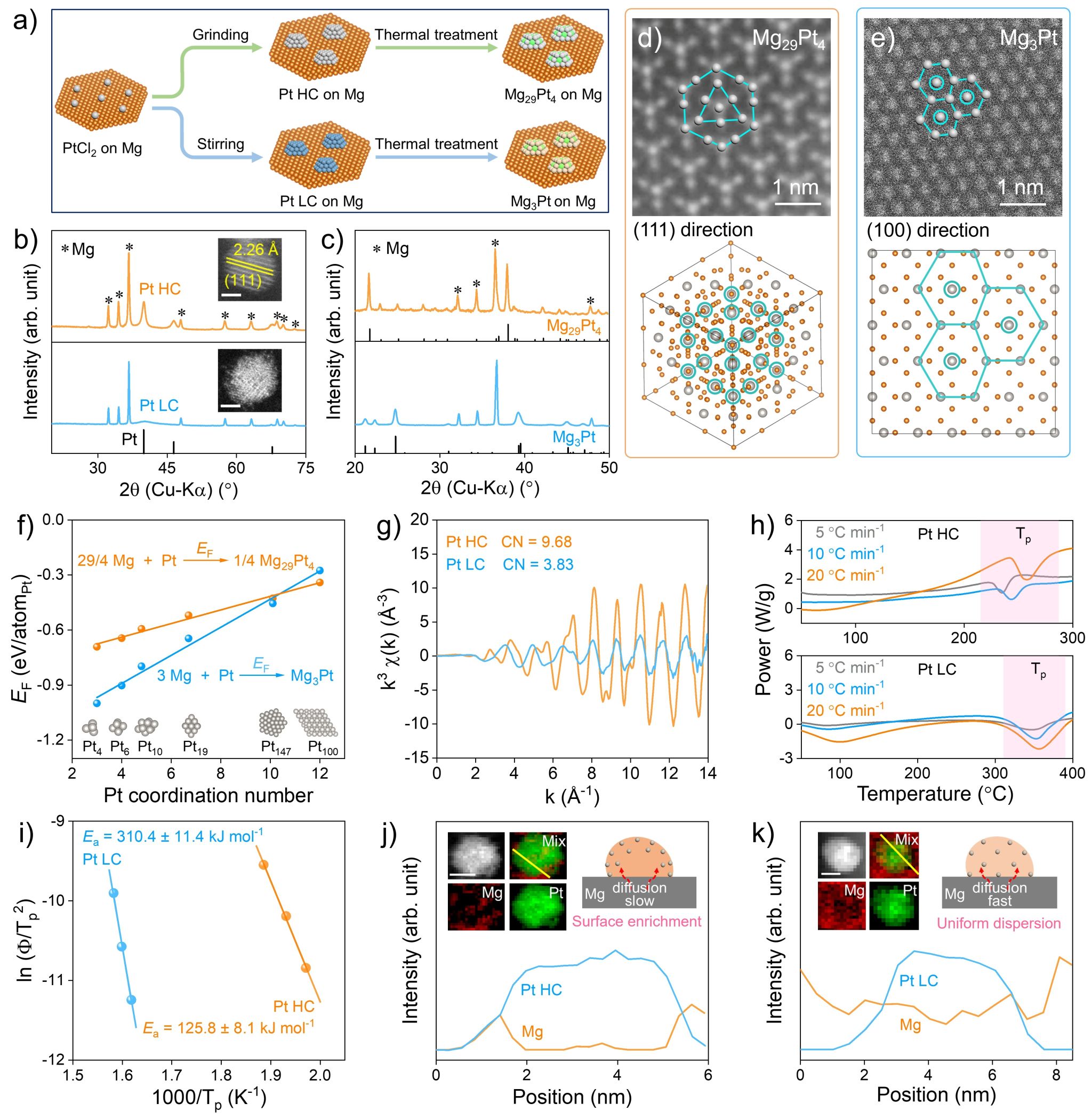
图2. Mg29Pt4与Mg3Pt的制备、表征以及形成机制。
如图2a所示,通过调控Mg基底表面Pt纳米颗粒(NPs)的结晶度,可以有效调节Mg与Pt NPs之间的反应性金属-载体相互作用(RMSI),进而实现Mg29Pt4与Mg3Pt两种材料的可控制备。如图2b,X射线衍射(XRD)与透射电子显微镜(TEM)结果证实了两种Pt NPs在结晶度上的差异:其中,通过研磨法制备的Pt NPs结晶度较高(简称为Pt HC),而采用搅拌法则得到结晶度较低的Pt NPs(简称为Pt LC)。接着,在相同的热处理条件下,Pt HC转变成Mg29Pt4,而Pt LC则转变成Mg3Pt。XRD与球差电镜结果证实了Mg29Pt4与Mg3Pt两种材料的成功制备(图2c-e)。
为了揭示Mg29Pt4与Mg3Pt的形成机制,作者计算了Mg29Pt4与Mg3Pt的热力学形成能(EF)随Pt配位数(CNPt)的变化关系。如图2f所示,Mg29Pt4与Mg3Pt两种材料的热力学稳定性与Pt的配位数密切有关。当Pt NPs的配位数较低时,Mg3Pt的热力学形成能低于Mg29Pt4,体系倾向于形成贫镁结构的Mg3Pt;而当Pt NPs的配位数较高时,Mg29Pt4表现出更高的热力学稳定性,此时更易形成富镁结构。图2g也证实了两种Pt NPs具有不同的配位数,其中Pt HC的配位数较高,而Pt LC的配位数较低,证实了理论计算的可靠性以及两种材料可控制备的可行性。
从动力学角度来看,两种Mg-Pt金属间化合物(IMCs)材料形成过程主要涉及Mg原子的扩散以及原子有序化排列,其难易程度可以通过形成过程的活化能(Ea)来反映。如图2h所示,作者利用差示扫量热(DSC)进行Ea值的测量(图2i)。其中,Pt HC和Pt LC形成对应IMCs的活化能分别为125.8 ± 8.1 kJ·mol-1与310.4 ± 11.4 kJ·mol-1,表明两种Mg-Pt IMCs的形成过程,具有不同的反应动力学特征。为了可视化该动力学过程,作者进一步采用电子能量损失谱(EELS)对材料的元素分布进行分析,以研究Mg原子在Pt NPs中的动态扩散过程。如图2j与2k所示,EELS面扫分析(EELS mapping)与EELS线扫分析结果表明:对于Pt HC样品,在加热过程中,Mg原子主要在Pt NPs的表面扩散并富集,形成类核-壳结构(图2j),有助于形成富镁结构的Mg29Pt4;而在Pt LC样品中,Mg原子均匀分布于Pt NPs的晶格内部(图2k),从而形成贫镁结构的Mg3Pt。
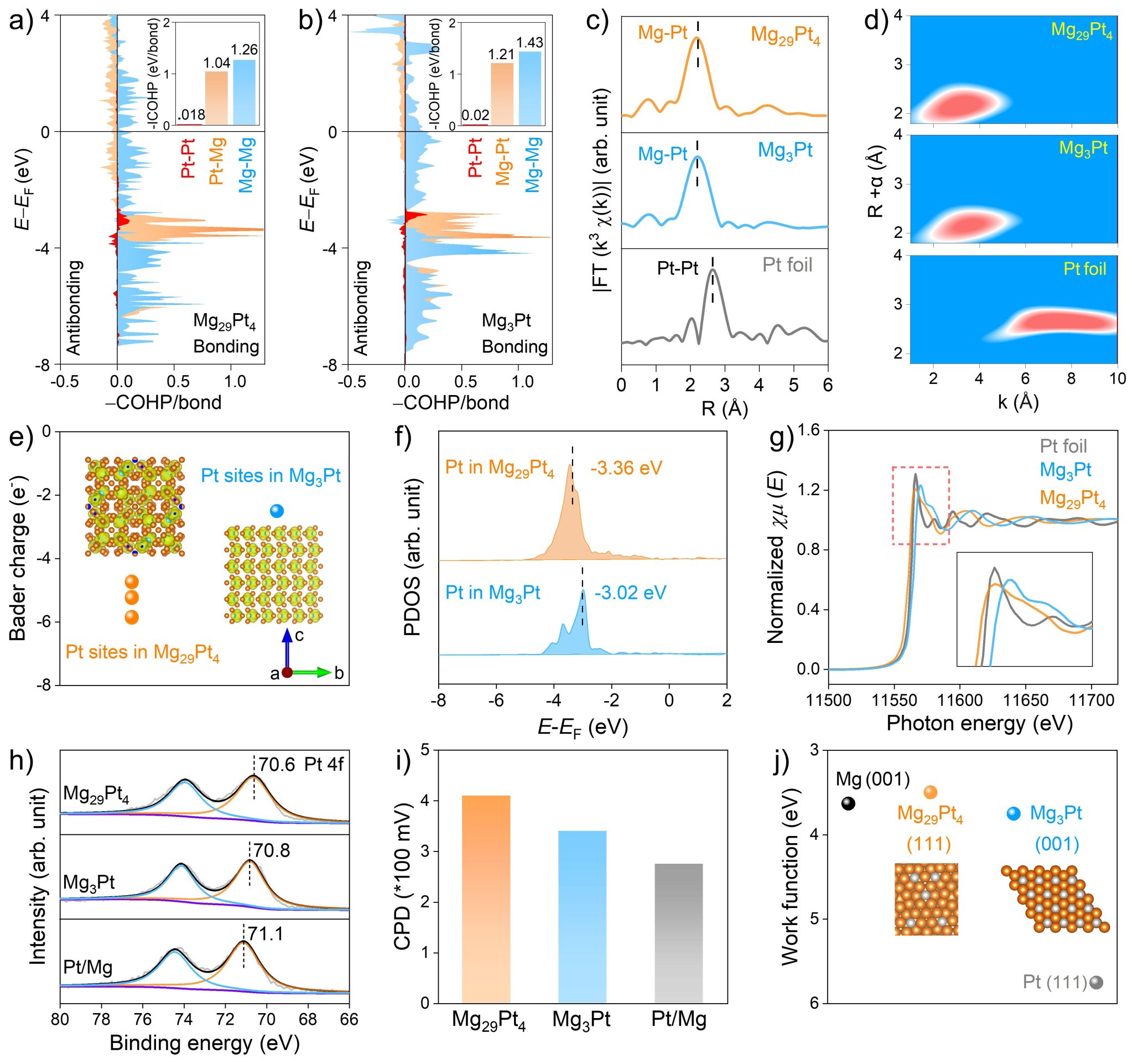
图3. Mg29Pt4与Mg3Pt的几何与电子结构表征。
作者对Mg29Pt4与Mg3Pt的几何与电子结构进行表征。如图3a与3b的晶体轨道哈密顿布局(COHP)所示,在Mg29Pt4与Mg3Pt材料中,活性Pt位点之间的-ICOHP值近乎为零,说明活性Pt原子之间几乎不存在电子相互作用,是以单原子的形式存在于晶格之中。此外,傅里叶变换的拓展边X射线吸收谱(FT-EXAFS)与小波变换的EXAFS谱图(WT-EXAFS)结果表明,在Mg29Pt4与Mg3Pt中,不存在Pt-Pt的配位特征峰,进一步证实了Pt原子是以单原子形式存在(图3c与3d)。上述表征结果说明,两种Mg-Pt IMCs均属于单原子金属间化合物材料(SAIMCs)。
接着,作者研究了Mg29Pt4与Mg3Pt中Pt位点的电子结构。如图3e所示,Bader电荷计算结果表明,与Mg3Pt相比,Mg29Pt4中Pt位点的电子密度更高。此外,X射线吸收近边结构(XANES)与X射线光电子能谱(XPS)也证实,Mg29Pt4和Mg3Pt中的Pt位点均带负电荷,但Mg29Pt4中Pt位点的电子密度更高(图3g与3h)。更高的电子密度,可以降低Mg29Pt4材料中Pt位点的d带中心。如图3f所示,在Mg29Pt4中,Pt位点的d带中心为-3.36 eV,明显低于Mg3Pt中Pt位点的d带中心(-3.02 eV)。通常,更高电子密度,可以抑制CO分子中5σ轨道电子向Pt的5d空轨道转移,而较低的d带中心,可以弱化Pt的5d电子反向填充至CO的2π*反键轨道,从而提高催化剂材料的抗CO中毒性能。
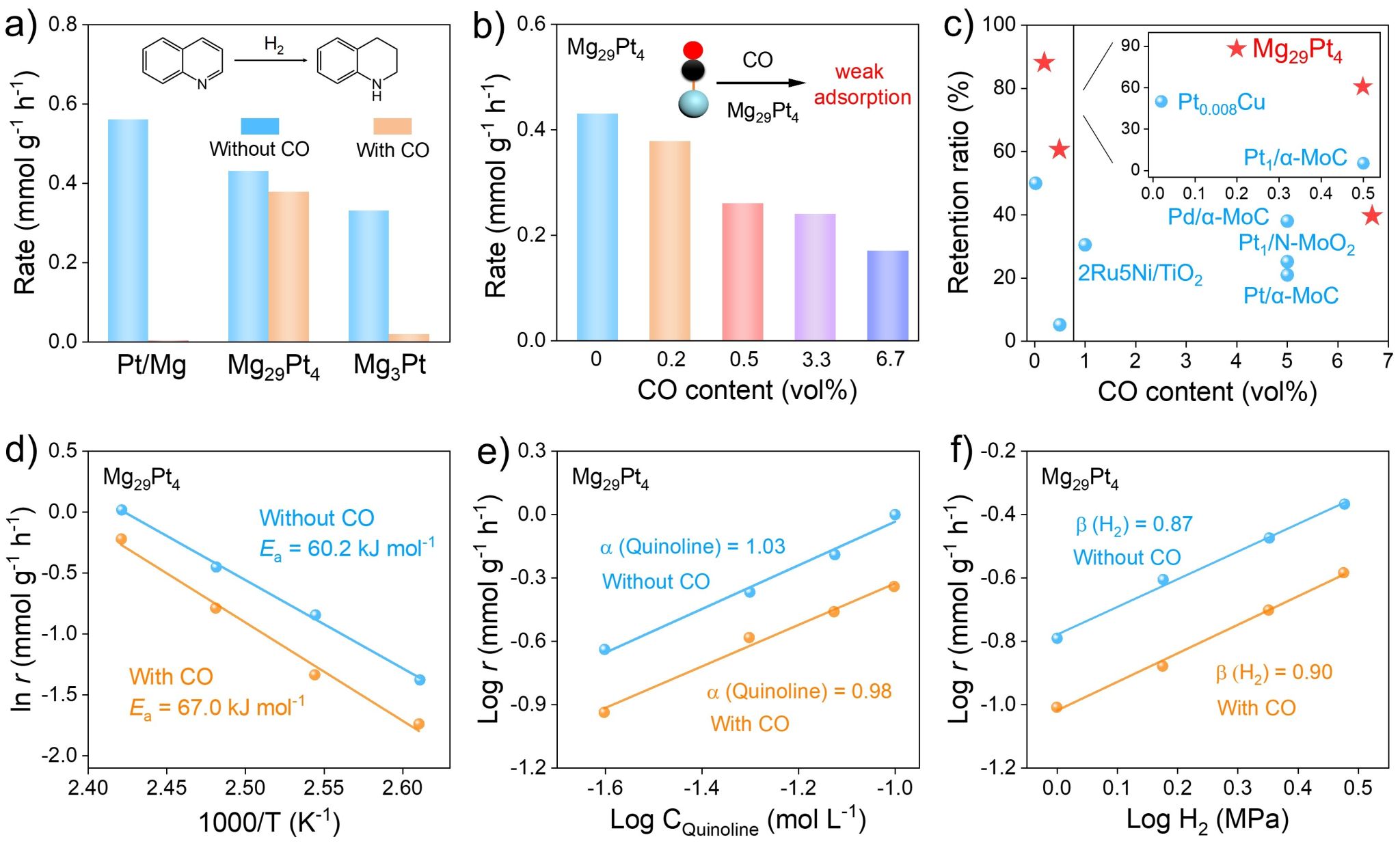
图4. 喹啉加氢抗CO中毒性能研究。
基于两种材料的结构特征,作者将Mg29Pt4与Mg3Pt应用于喹啉加氢抗CO中毒反应中。如图4a所示,当在H2体系中引入0.2 vol% CO时,Pt/Mg催化剂发生了严重的CO中毒现象。相比之下,当Mg与Pt NPs形成Mg-Pt IMCs后,Mg29Pt4和Mg3Pt催化剂表现出CO耐受性。其中,Mg29Pt4的加氢抗CO催化活性较Mg3Pt催化剂提升了21倍。此外,与纯H2体系相比,Mg29Pt4催化剂的抗CO加氢活性保留率可高达88%,展现出优异的抗CO毒化能力。为进一步评估Mg29Pt4催化剂的抗CO中毒能力,作者研究了H2中的CO浓度对反应速率的影响。如图4b与4c所示,即使CO浓度增加到6.7 vol%,Mg29Pt4催化剂仍然能够保留约40%的初始活性。
接着,作者研究CO引入对催化反应动力学的影响。如图4d-f所示,在0.5 vol% CO/H2条件下,Mg29Pt4催化喹啉加氢反应的活化能、喹啉与H2的反应级数,和纯H2条件下相近,表明CO的加入并不会改变Mg29Pt4的催化反应动力学过程,进一步证实了Mg29Pt4催化剂具有优异的抗CO毒化能力。
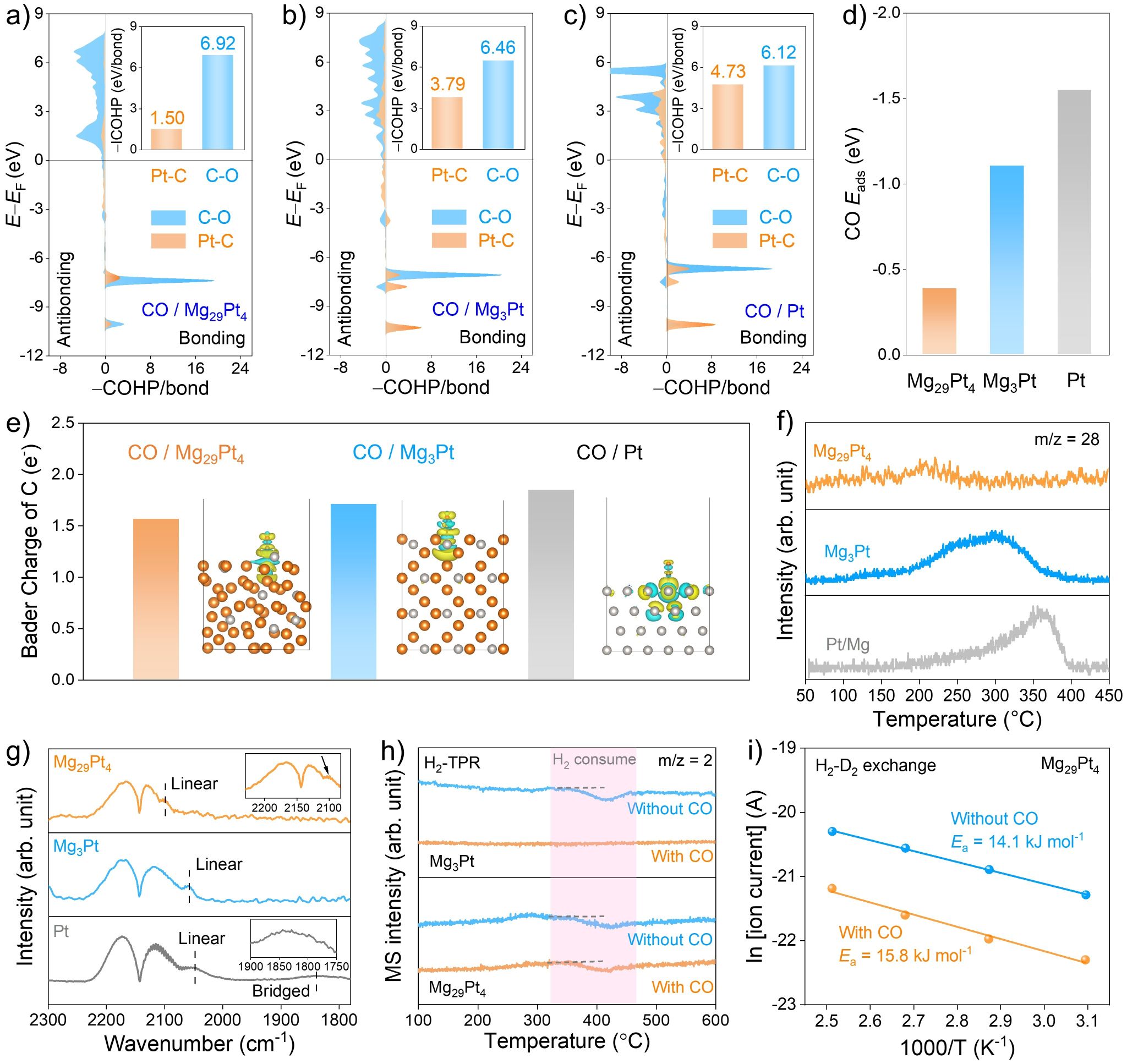
图5. 抗CO中毒的机理研究。
为了阐明Mg29Pt4与Mg3Pt两种催化剂抗CO毒化能力的差异,作者通过COHP计算研究了Mg29Pt4、Mg3Pt和Pt表面吸附的CO分子中C-O化学键的强度以及Pt-C化学键的强度。如图5a-c所示,在Mg29Pt4上,CO分子与Pt位点之间Pt-C化学键的-ICOHP值为1.50 eV/bond,远远低于Mg3Pt与纯Pt,表明CO与Mg29Pt4中Pt位点之间相互作用较弱。同时,Mg29Pt4表面吸附的CO分子中C-O化学键的-ICOHP值为6.92 eV/bond,高于Mg3Pt和纯Pt,说明CO在Mg29Pt4表面的活化程度较低,进一步验证了Mg29Pt4材料与CO分子之间的弱相互作用。此外,CO吸附能计算结果表明,Mg29Pt4上CO的吸附能远低于Mg3Pt和纯Pt(图5d)。以上结果充分证实Mg29Pt4催化剂具有优异的抗CO毒化能力。
实验上,作者首先采用CO的程序升温脱附(CO-TPD)技术分析了CO分子在Pt、Mg3Pt和Mg29Pt4表面的吸/脱附行为。如图5f所示,CO分子在Mg29Pt4催化剂上的吸附强度最弱,并且脱附温度最低,表明Mg29Pt4与CO之间的相互作用最弱。此外,作者通过原位漫反射红外傅里叶变换光谱(DRIFTS)进一步研究了催化剂的CO吸附能力。如图5g所示,CO分子在Mg29Pt4表面线性吸附模式所对应的振动频率高于Mg3Pt和Pt/Mg,进一步证实其更优异的抗CO中毒特性。
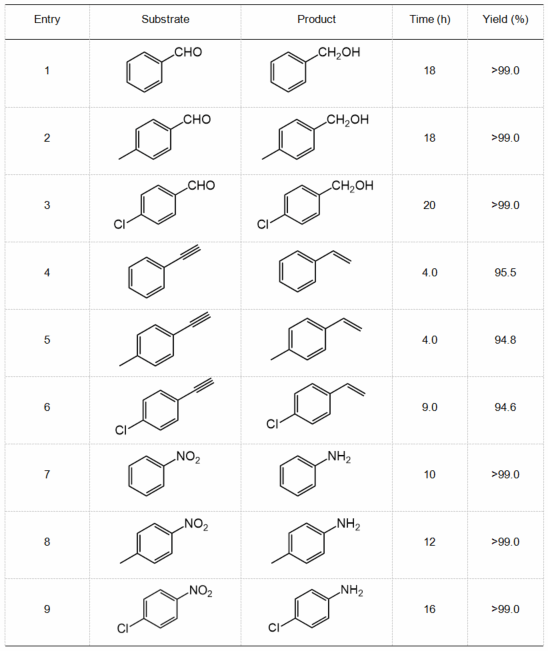
为研究CO引入对催化剂表面H2解离行为的影响,作者进行了H2程序升温还原(H2-TPR)测试与H2-D2交换实验。如图5h所示,当向体系引入CO后,Mg29Pt4催化剂上H2解离峰几乎保持不变,表明CO的存在并未改变催化剂表面的H2解离行为。此外,H2-D2交换实验结果表明,在CO存在条件下,Mg29Pt4催化H2-D2交换的表观活化能与纯H2条件下的测定值相近,进一步证明Mg29Pt4催化剂具备优异的加氢抗CO中毒能力(图5i)。将反应体系拓展至其它底物分子,例如含有炔基、醛基及硝基官能团的反应物,在含CO的氢气中,Mg29Pt4催化剂仍表现出高的加氢活性与选择性,展现出广泛的底物适用性。
表1. 底物拓展
综上研究,通过控制Mg基底表面Pt NPs的结晶度,可以有效调节Mg与Pt NPs之间的RMSI,从而实现Mg29Pt4和Mg3Pt两种不同晶体结构SAIMCs材料的可控制备。其中,Pt位点均以单原子形式有序分布于对应的晶体结构之中,但其电子结构存在显著差异,从而导致二者在加氢抗CO毒化反应中表现出不同的催化性能。在含有0.2 vol% CO的喹啉加氢体系中,Mg29Pt4的催化活性较Mg3Pt催化剂提升了21倍。此外,相比于纯H2体系,Mg29Pt4催化剂的抗CO加氢活性保留率高达88%,表现出优异的抗CO中毒性能。机理研究表明,Mg29Pt4催化剂优异的抗CO毒化性能主要源于其Pt位点具有较高的电子密度和较低的d带中心。这一独特的电子结构能有效抑制CO 5σ轨道电子向Pt 5d轨道的填充以及Pt 5d电子向CO 2π*反键轨道的填充,从而显著弱化CO在Pt位点上的吸附,最终提升Mg29Pt4催化剂的加氢抗CO毒化能力。此外,Mg29Pt4催化剂可成功应用于多种有机官能团(如醛基、硝基以及炔烃基团)的抗CO加氢,具有广泛的底物适用性。
原文链接:https://onlinelibrary.wiley.com/doi/epdf/10.1002/anie.202516790
导师介绍
叶天南,上海交通大学变革性分子前沿科学中心课题组组长 (PI),长聘教轨副教授,博士生导师。主持面上项目、青年项目、日本学术振兴会JSPS海外项目,承担国家重点研发项目子课题,上海市科技重大专项子课题,入选上海市领军人才(青年)计划、上海市浦江人才计划,上海市教委人工智能跃升计划,入选2025全球前2%顶尖科学家年度科学影响力榜单,获得美国化学会ACS Catalysis青年研究员奖。2015年在上海交通大学获得博士学位,同年进入日本东京工业大学元素战略研究中心任特任助理教授,期间担任日本学术振兴会特别研究员 (JSPS fellow),迄今,共发表研究论文60余篇,其中第一作者/通讯作者论文37篇,代表性论文包括Nature,Nat. Catal.,Nat. Commun.,J. Am. Chem. Soc.,Angew. Chem. Int. Ed.,Adv. Mater.等,获授权中国发明专利12项,美国专利1项。担任国际知名期刊Progress in Reaction Kinetics and Mechanism副主编,Crystal杂志客座编辑,中国化学会Chem. Res. Chin. 和Chin. Chem. Lett.期刊青年编委。
课题组尚有2026年春季/秋季入学“申请-考核”制博士生名额,欢迎具有无机化学与功能材料、催化与表界面化学、高温电化学等相关背景的同学加入我们团队。请有意者发送简历和研究小结至ytn2011@sjtu.edu.cn。同时,课题组长期招聘无机合成化学、无机功能材料、催化与表界面化学以及高温电化学等相关领域的博士后,待遇从优,欢迎感兴趣的同学随时邮件联系。




