

科学研究


近日,上海交通大学吴晶晶团队在CCS Chemistry上发表了题为“Bioinspired Schenck-ene/Hock/aldol cascade reaction Enables Concise Synthesis of Natural Products Alstoscholarinoid A, Masterpenoid D, Leontogenin and Marsformoxide B”的研究论文。甾体与萜类天然产物根据其核心分子骨架的差异可划分为不同的家族,这些核心骨架往往复杂多变,彼此间差异明显。通常天然产物合成化学家需要针对不同的核心骨架设计相应的天然产物合成策略。因此,以相同或相似的策略集群式合成(collective synthesis)具有不同分子骨架的天然产物对于简化天然产物合成策略及提高合成效率具有重大意义。天然产物alstoscholarinoid A(1)、masterpenoid D(2)、leontogenin(3)和marsformoxide B(4)分属三种不同的家族,具有多种生理活性(图 1)。上海交通大学吴晶晶团队与中国科学院上海有机化学研究所薛小松团队合作,报道了一种单线态氧气分子介导的新型仿生Schenck-ene/Hock/aldol串联重排反应的分子骨架编辑(skeletal editing)策略。他们不仅利用该关键策略快速完成了天然产物1~4的合成,还通过进一步的底物扩展,验证了该串联反应策略对不同分子进行骨架编辑的有效性。此外,他们还结合DFT理论计算,进一步深入探究了该串联反应的机理和控制因素。
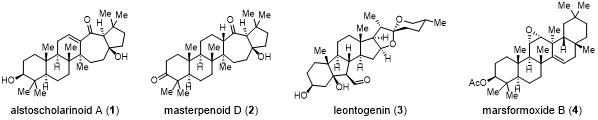
图 1 天然产物alstoscholarinoid A、masterpenoid D、leontogenin和marsformoxide B
作者的研究始于对alstoscholarinoid A(1)的合成。根据生源合成假说,1的双环[5.3.0]癸烷骨架是1,6-环癸二酮底物(6)经分子内跨环aldol反应得到的(图 2A)。根据该生源合成假说,他们从齐墩果酸(5)出发,经过6步反应合成了相应的跨环aldol反应前体8。8在各类反应条件下主要生成产物9~11(图 2B)。X射线晶体衍射实验(XRD)进一步表明具有8的1,6-环癸二酮骨架具有两种不同的构象,而9和10的立体化学可能受这两种构象诱导与控制(图 2C)。鉴于8的跨环aldol反应主要经C16位烯醇化得到9~11而非C19位烯醇化得到1的分子骨架,作者设计了一种迂回的合成策略以获得C19位烯醇化的中间体。从8出发,经选择性保护C17位羰基与羰基烯醇化反应,可以得到C19位烯醇化的12。然而12对各种反应条件非常敏感,在进一步氧化修饰C19位或仅仅脱除1,3-二硫戊环烷保护基时常常剧烈分解。经过反复尝试,他们最终在12与N-氯代丁二酰亚胺于低温下快速反应后得到13。此时只要完成13中四取代不饱和酮的水合反应及去乙酰化反应就能得到1。然而经过尝试后,作者发现水合反应难以发生(图 2B)。这表明直接通过分子内跨环aldol反应合成1的策略难以继续进行下去,需要重新设计于优化路线。
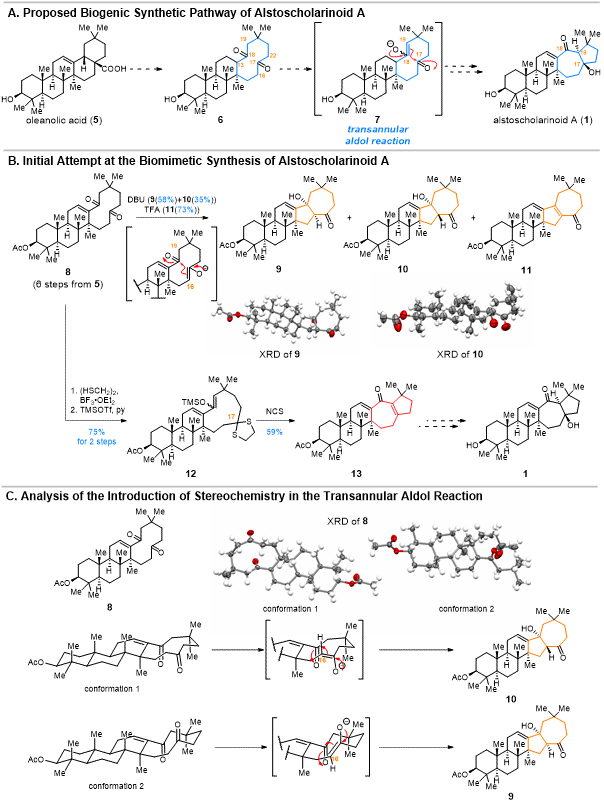
图 2 基于跨环aldol反应的Alstoscholarinoid A(1)的仿生合成研究
第一阶段研究结果表明跨环aldol反应的难点在于控制底物在C19位发生烯醇化。受Pak课题组合成clavukerin C的启发,作者提出从共轭二烯底物14出发经Schenck-ene反应和酸催化Hock反应可制得C19位烯醇化的中间体15,而该中间体再经aldol反应有望生成1。新策略有望从14出发经一锅(one-pot)反应得到1,可大幅提高合成的简洁度与效率(图 3A)。在初步实验中14与单线态氧气分子反应可以得到过氧烯丙醇16,而16与催化量的质子酸作用或仅仅溶解在氘代氯仿(通常含有微量HCl)就可转化为19(乙酰化的1)。这表明作者的设想是合理的。为了探究该反应的机理,需要进一步确定关键中间体16的立体结构。然而16的稳定性差,难以进行深入的结构表征研究,因此作者采取了各种间接方法鉴定其结构。他们首先通过还原反应得到烯丙醇12表明16存在过氧醇基团。在以四苯基卟啉(TPP)为光敏剂、CCl4为溶剂中,14经Schenck-ene反应得到另一个中间体20,该化合物稳定性较高,可通过XRD表征其结构。观察20的结构,可知其六元环内过氧化结构是16中的S-cis共轭二烯与单线态氧气分子经[4+2]
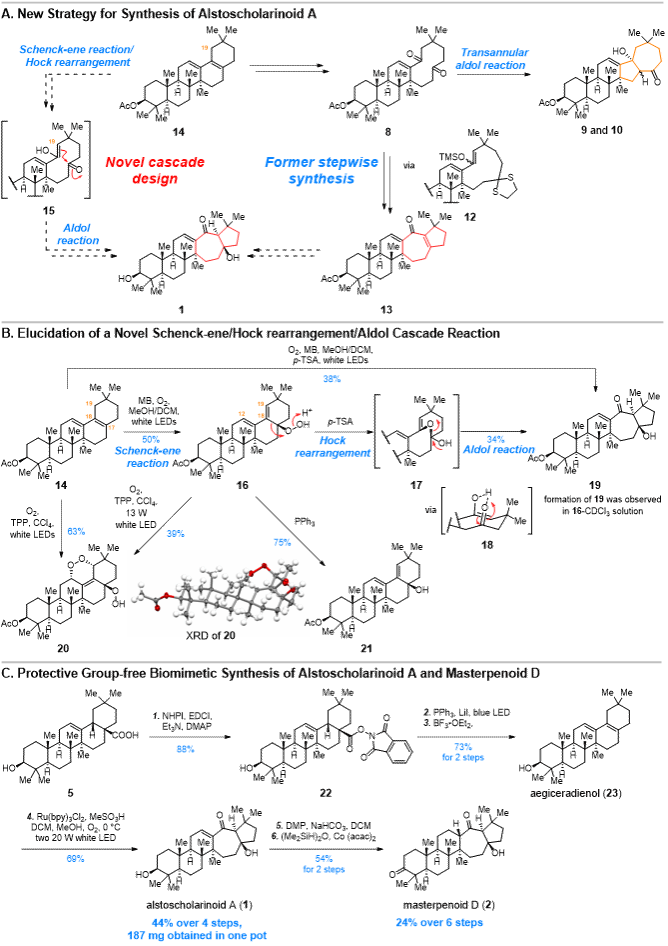
图3 仿生Schenck-ene/Hock/aldol串联重排反应的发现与alstoscholarinoid A(1)和masterpenoid D(2)的合成
环加成反应得到的。因此20的生成不仅间接证明了16的存在,还可通过立体化学传递原理推断出16中过氧醇的立体构型与作者推断的一致。进一步研究也表明16在TPP/CCl4/O2体系中也生成了20,而14在酸催化下经与单线态氧气分子反应后也可直接得到19。基于上述实验结果,作者初步认为该串联重排反应是Schenck-ene/Hock/aldol过程,其中16经Hock重排得到氧桥中间体17,再经分子内重排反应得到烯醇18,而aldol反应的立体化学则受18中分子内氢键的控制(图 3B)。在拓展研究中,作者发现底物中的游离羟基不受反应影响,因此改以天然产物aegiceradienol(23)优化该串联反应,并将最优反应条件用于1的合成研究。最终他们以齐墩果酸(5)为原料经4步反应,实现了1的百毫克级无保护基仿生合成,总收率达44%,且从1出发经两步反应制得天然产物masterpenoid D(2,图 3C)。根据上述结果,作者也提出天然产物1和2的另一种生源合成假说,即从23出发,经历一种非酶催化的串联Schenck-ene/Hock/aldol反应过程。
近年来分子骨架编辑策略在天然产物合成与结构修饰上的重要性有目共睹。为此作者决定发掘Schenck-ene/Hock/aldol串联重排反应在骨架编辑中的潜力。首先作者通过合成1的衍生物24b~27b表明该重排反应条件温和,官能团容忍性高,一些对酸敏感的官能团如肟、MOM都能兼容(图4A)。对于三取代与四取代烯烃底物,TPP/CCl4体系的效率更高。在该体系下Δ5-甾体底物28a~30a可分别转化为abeo-5(6→7)产物28b, 29b和天然产物leontogenin(3),而Δ9,10-八氢化萘底物31a~33a也可分别转化为具有双环[5.3.0]癸烷骨架的共轭不饱和酮产物31b~33b,其中32b和33b还具有天然产物中常见的顺式双环[3.3.0]辛烷骨架(图4B)。这些结果充分表明Schenck-ene/Hock/aldol串联重排反应是一种新型的骨架编辑手段。然而,进一步研究表明,并非所有过氧烯丙醇都会发生该串联重排反应。如根据文献报道β-香树脂醇醋酸酯34经Schenck-ene反应后可生成中间体35。然而作者发现34在常规条件下难以生成35,最终经过自由基烯丙基C-H键氧化反应才得到35。在质子酸催化下35未能发生串联重排反应,而是经oleanane→taraxastane重排反应得到另一个天然产物marsformoxide B(4,图 4C)。

图4 基于Schenck-ene/Hock/aldol串联重排反应的骨架编辑研究
尽管作者已经通过实验手段深入研究了Schenck-ene/Hock/aldol串联重排反应的机理,但该反应机理的细节,如氧桥中间体17是否合理、aldol反应的立体化学是否受18中分子内氢键的控制及何种因素控制过氧键迁移的结果等依然有待研究。为此作者与中国科学院上海有机化学研究所的薛小松团队合作,通过密度泛函理论(DFT)对反应机制进行计算化学研究。在16→19的反应中,作者以对甲基苯磺酸(p-TSA)为催化剂、CH2Cl2和MeOH为溶剂。计算结果显示16经Hock重排反应(过渡态为TS-A)生成17的吉布斯自由能垒为20.1 kcal/mol,表明该重排可在273K下进行。在TS-A中p-TSA通过与过氧醇的羟基形成氢键作用促进过氧键的断裂与新C-O键的形成。在后续反应中17的氧桥基团在p-TSA的作用下发生碎裂反应生成18'(能垒为7.5 kcal/mol),最后18'经aldol反应生成19(能垒为7.2 kcal/mol)。在TS-B和TS-C中,p-TSA通过质子穿梭作用(proton shuttle)促进反应进行。反应的总吉布斯自由能变化为−61.6 kcal/mol,是一个强烈放热的过程(图 5A)。
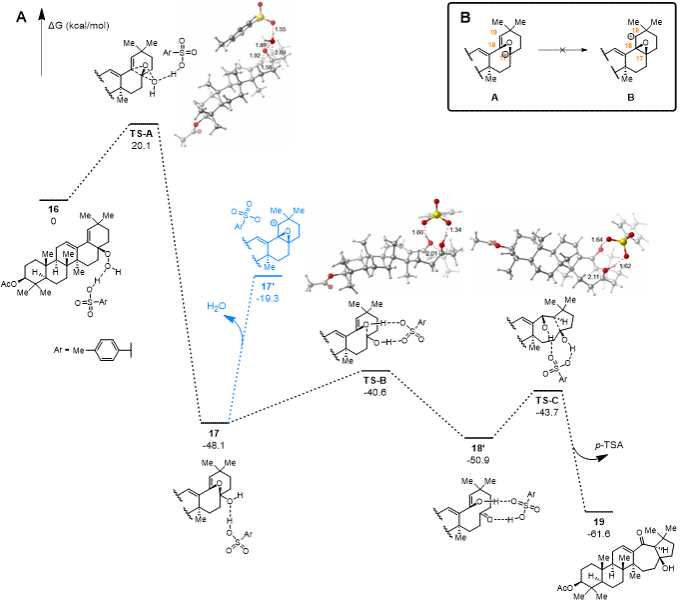
图5 化合物16→19转化的吉布斯自由能变化图
随后,作者评估了35→4的转化,并以MeSO3H为催化剂。计算结果表明35未能生成任何Hock重排的中间体,而是首先经过渡态TS-D生成α-环氧碳正离子IM-1。该反应的吉布斯自由能垒为18.1 kcal/mol,表明该重排同样可在273K下进行。IM-1经过渡态TS-E生成甲基迁移的碳正离子IM-2的能垒为5.8 kcal/mol,而经过渡态TS-E2生成类似Hock重排的扩环碳正离子IM-2'的能垒为8.2 kcal/mol。因此IM-1朝着能垒更低的方向转化为IM-2,最终转化为4。整个反应的总吉布斯自由能变化为−53.8 kcal/mol(Scheme 5A)。作者进一步通过比较碳正离子A、B、C和D(图 5B和6B)的相对稳定性来探讨影响过氧烯丙醇后续转化途径的因素。他们认为主要有两个因素影响这些中间体的相对能量:环氧基团的张力和碳正离子的稳定性。B中的四取代环氧基团(对应图 5A中17')位于两个环中央,其张力高于C中的cis-1,2-二取代环氧基团。C中的三级碳正离子稳定性也高于B中的二级碳正离子,而A中的二级碳正离子稳定性也高于D中的一级碳正离子。由此可知在图 5B中B因稳定性较差而难以生成,而图 6B中C因环氧张力相对较低且碳正离子相对稳定而更易生成。因此,TS-A倾向于生成氧桥中间体17,然后发生Hock/aldol串联反应,而TS-D生成的环氧中间体IM-1在动力学上倾向于形成IM-2,然后进一步生成4。这就解释了在这两种情况不同的底物中观察到的不同选择性。

图6化合物35→4转化的吉布斯自由能变化图
至此,作者通过发展一种新型仿生Schenck-ene/Hock/aldol串联重排反应,从廉价易得的天然分子出发以1~6步反应集群式合成了四种天然产物。该串联反应条件温和、官能团容忍性高,可作为天然产物分子骨架编辑的新策略。该研究成果近日以“Bioinspired Schenck-ene/Hock/aldol cascade reaction Enables Concise Synthesis of Natural Products Alstoscholarinoid A, Masterpenoid D, Leontogenin and Marsformoxide B”为题,在国际著名期刊CCS Chemistry上线发表(DOI: 10.31635/ccschem.025.202506037)。上海交通大学李若曦博士作为论文第一作者,负责本文的合成研究,中国科学院上海有机化学研究所的王桐坤博士作为论文共同第一作者,负责本文的计算化学研究,指导老师为上海交通大学吴晶晶副教授和上海有机化学研究所的薛小松研究员。作者特别感谢上海有机所的田伟生研究员、桂敬汉研究员和以及英国Bristol大学的Varinder K. Aggarwal教授所提供的帮助。值得一提的是,在本文投稿其他期刊期间,维也纳工业大学的Kratena课题组也发表了他们以Schenck-ene/Hock/aldol串联重排反应合成alstoscholarinoid A的研究工作(JACS Au 2025, 5, 1076–1082)。




