

科学研究


近日,上海交通大学李俊团队在Journal of the American Chemical Society上发表了题为“Enduring CO Electrolysis with Ampere-Level Reaction Rates Using Nickel-Doped Iridium Catalysts”的研究论文。该研究开发了一种在氧化铱载体中低浓度掺杂镍的阳极催化剂,显著增强了铱的结构稳定性,并有效抑制了镍位点的氧化。所构建的低价态镍掺杂氧化铱阳极材料在对液体产物保持良好氧化惰性的同时,兼具优异的析氧性能与长期稳定性。原位软X射线光电子能谱表征表明,该催化剂的活性中心主要由Ni2+和Ir4+组成。将其集成于膜电极组件(MEA)系统中,可在Cu阴极条件下实现高效且稳定的CO电还原反应,在1 A/cm2电流密度下达到32%的全电池能量效率(EE)和73%的单程碳利用效率(SPCE),并可持续稳定运行超过1000小时。

CO2/CORR在可再生电力驱动下,为将捕集的CO2转化为高附加值多碳(C2+)产物提供了极具拓展潜力的路径。当前CO2/CORR技术已能以超过1000 mA/cm2的电流密度合成如乙烯、乙醇和正丙醇等C2+产物。然而,工业化面临的核心挑战在于同时实现高FE%、足够的EE%以降低运行成本,以及高SPCE以减少后处理成本。MEA电解槽体系被广泛认为是实现CO2/CORR工业化的理想架构。近年来,中性条件下的MEA-CO₂RR已实现26%的EEC2+,但受限于盐析问题,其运行稳定性难以维持。改用纯水电解质虽可有效避免析盐,但显著牺牲了EE与SPCE。相比之下,级联电解策略中,CO2首先被还原为CO,随后在碱性环境中进一步转化为C2+产物,展现出同步提升EEC2+与SPCE的潜力。目前,将CO2转化为CO的电解技术在安培级电流密度下已具备优异的EE、SPCE和长期稳定性,因此研究重心已逐步转向后续的CORR过程优化。MEA-CORR体系在阴极材料与界面设计方面已实现1 A cm⁻²下32% EEC2+和17% SPCEC2+。但延长稳定性至≥500 h常导致性能下降。工业化关键在于打破EEC2+、SPCEC2+与稳定性之间的权衡。
在碱性MEA-CORR体系中,尽管初始FEC2+与EEC2+较高,但在高电流密度下,阳极侧pH显著下降,促使Ni或IrO2阳极发生溶解,所释放的金属离子迁移至阴极Cu催化剂表面,导致抑制CORR并促进析氢反应,导致阴极中毒,进而显著削弱体系的EEC2+与运行稳定性。同时,长时间运行导致CORR乙醇产物积累并向阳极扩散,在阳极氧化作用下进一步消耗产物,致使FEC2+下降(图1)。在MEA体系中,为实现高效OER而避免乙醇氧化,本工作设计了在IrO2表面负载低价Ni2+的小尺寸孤立结构。相比于传统NiOx掺Ir策略中Ni2+易被氧化为促进乙醇氧化的Ni3+,该结构有望稳定Ni价态,提升OER而抑制乙醇氧化。
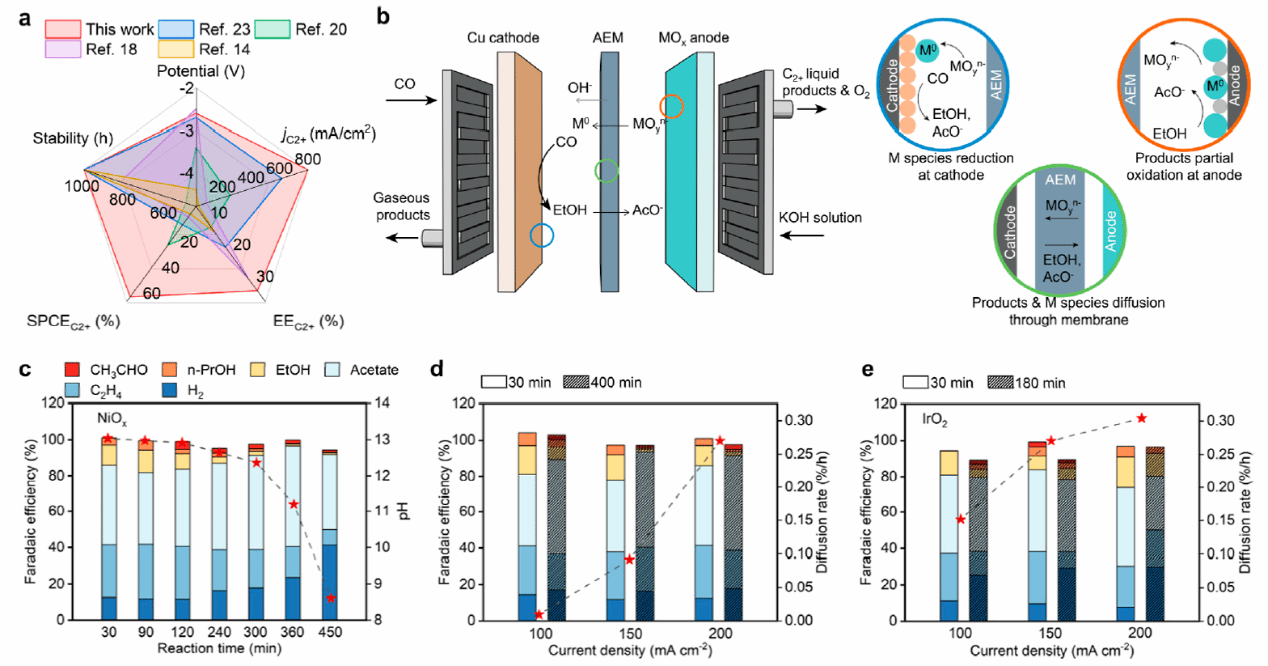
图1. (a)不同MEA-CO₂/CORR体系的性能对比(稳定性>500小时);(b) 传统阳极催化剂溶解过程和产物氧化过程示意图;(c) 镍基催化剂为阳极时,CORR电解过程中的性能与pH变化随反应时间的关系;(d) 镍基催化剂降解下的产物变化与阳极物种扩散; (e) IrO₂催化剂降解下的产物变化与阳极物种扩散。
为了深入了解反应机理,本工作进行了DFT计算(图2)。我们构建了Ni2+掺杂IrO2(LVN-IrOx)模型,分别计算其Pourbaix图与反应自由能。结果表明,LVN-IrOx在碱性高电位下结构稳定,优于易溶解的IrO2。对乙醇氧化反应,LVN-IrOx呈现较高反应势垒(ΔG = 1.68 eV),而NiOx则倾向于生成乙酸(ΔG = 1.11 eV)。在OER路径中,LVN-IrOx关键步骤的自由能最低(2.21 eV),并在Ir⁴⁺位点上表现出较低的*OH吸附能(0.51 eV),说明Ir⁴⁺对OER活性至关重要。
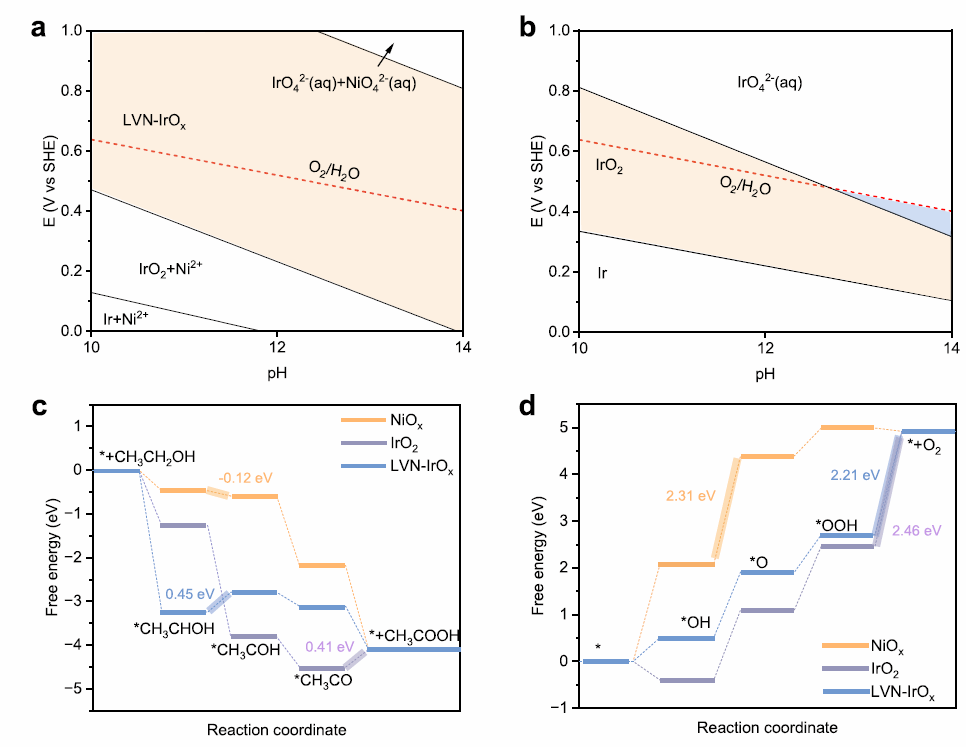
图2. (a) LVN-IrOx的Pourbaix图。(b) IrO₂的Pourbaix图。(c) 1.23 V时乙醇氧化反应在不同催化剂表面的自由能比较。(d) 0 V时OER反应在不同催化剂表面的自由能比较。
为制备稳定的 LVN-IrOx 催化剂,本工作将IrCl4溶于极性溶剂后浸涂于泡沫镍上,并通过退火实现Ni在IrO2基体中的原位掺杂。扫描电子显微镜(SEM)与透射电镜(TEM)显示形成了约150 nm的LVN-IrOx结构。XRD表明IrO₂晶面发生轻微衍射角偏移,表明Ni掺杂引起的晶格收缩。XAS/XANES/EXAFS共同验证Ir−Ni配位构建成功,配位数分析表明Ir−O约为6、Ir−Ni约为4,小波变换亦印证Ir−O−Ni局域结构特征(图3)。
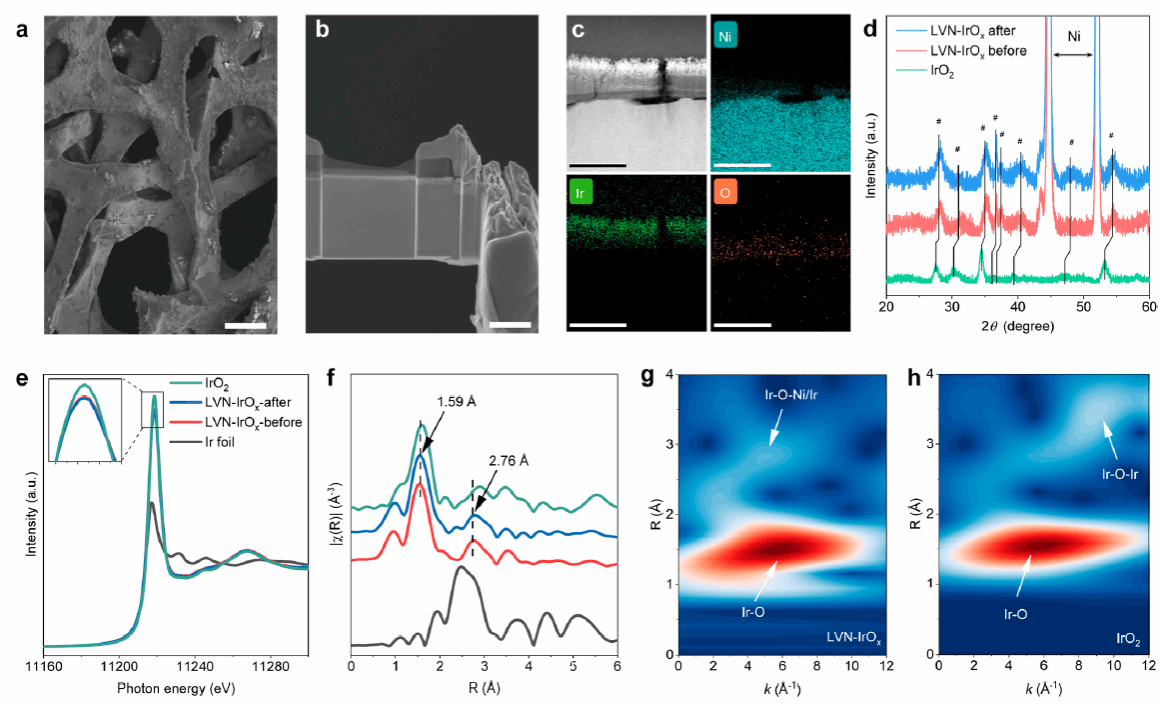
图3 (a) 催化剂在泡沫镍上的SEM形貌图;(b, c) 催化剂层截面结构的及元素分布;(d) 电解前后催化剂的XRD图谱;(e, f) 电解前后 Ir L3-edge的XANES与EXAFS光谱;(g, h) LVN-IrOx和IrO2电解后 Ir L3-edge的小波变换图。
为揭示LVN-IrOx在电解过程中的电子结构特征,采用原位近常压X射线光电子能谱 (NAP-XPS)对其进行分析(图4a)。结果显示,在开路电压下,Ni主要以低价态Ni²⁺存在,Ir则表现为Ir3+/Ir4+混合态,且Ni2+在0.8–2.3 V电位区间内保持稳定,而Ir3+逐渐氧化为Ir4+,表明Ir4+是OER的主要活性中心(图4b)。O 1s谱分析进一步表明,随着电位升高,OH逐步转化为OOH,有助于O₂生成,与DFT计算结果一致(图4c)。此外,原位拉曼测试表明,LVN-IrOx对乙醇氧化反应表现出惰性,电位升高过程中乙醇和水的特征峰基本无变化(图4d)。而NiOx则表现出乙醇峰位红移和水峰位蓝移,且在925 cm-1处出现乙酸中*CH3基的特征峰(图4e和4f)。
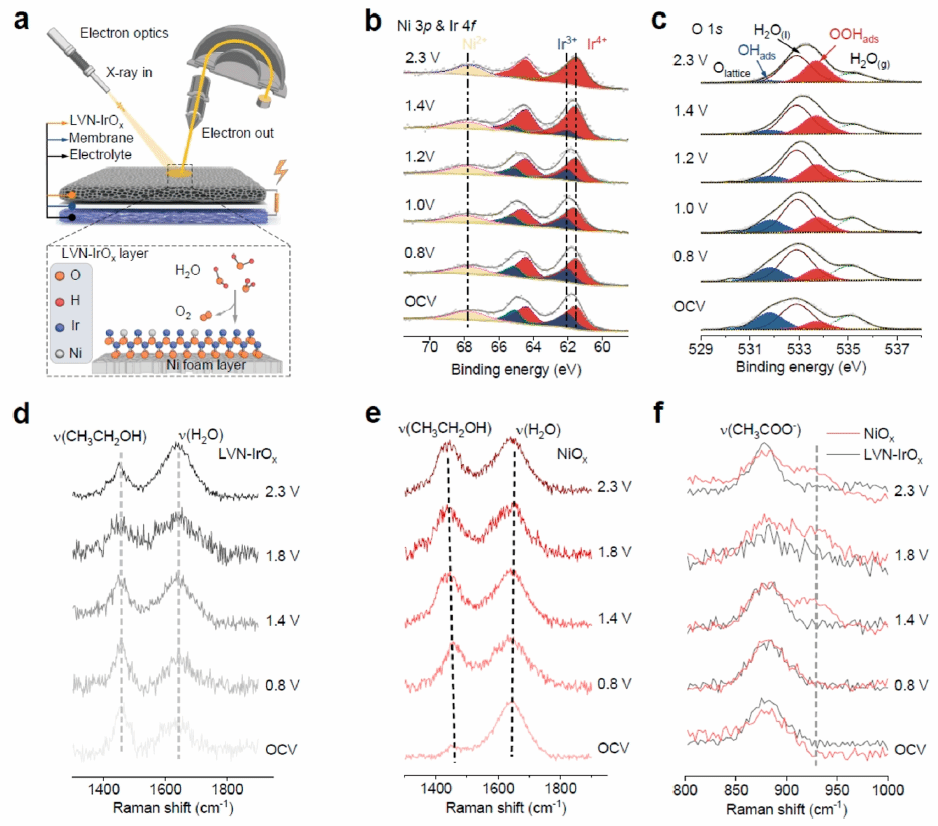
图4 (a)原位电化学NAP-XPS装置示意图。(b) OER过程中,不同电位下Ni 3p与Ir 4f的原位NAP-XPS谱图。(c) OER过程中O 1s原位NAP-XPS谱图。(d) LVN-IrOx催化剂在不同电位下乙醇电化学氧化过程中的原位拉曼光谱。(e) NiOx催化剂在相同条件下的原位拉曼光谱。(f) LVN-IrOx与NiOx在800-1000 cm-1波数范围内的原位拉曼谱对比图。
最后,本研究测试了LVN-IrOx在1 M KOH电解质中的性能。与NiOx和IrO₂相比,LVN-IrOx展现出更高的电流密度(约1200 mA cm-2),且在相同电流下降低了0.5 V的槽压,显著提升了EEC2+(图5a和5b)。槽压拆分结果显示,LVN-IrOx的阳极过电位仅为180 mV,明显优于IrO₂(580 mV),性能提升主要归因于Ni²⁺对Ir⁴⁺活性的增强而非仅仅提高催化剂电化学活性面积(图5c)。
在200mAcm-2下运行120 h,LVN-IrOx仍保持80%的FEC2+与34.2%的EEC2+,显示出优异的稳定性(图5d和5e)。相比之下,IrO₂和NiOx阳极体系在24 h后性能明显下降。进一步在1000 mA cm-2下运行时,配合多孔铜阴极,LVN-IrOx实现了超过80%的FEC2+与高达45%的EEC2+。当CO进气速率降低时,SPCEC2+最高提升至73%。在1000 mA cm-2下连续运行1000 h,系统仍保持约80%的FEC2+与32%的EEC2+,展现出显著的结构稳定性(图5f)。
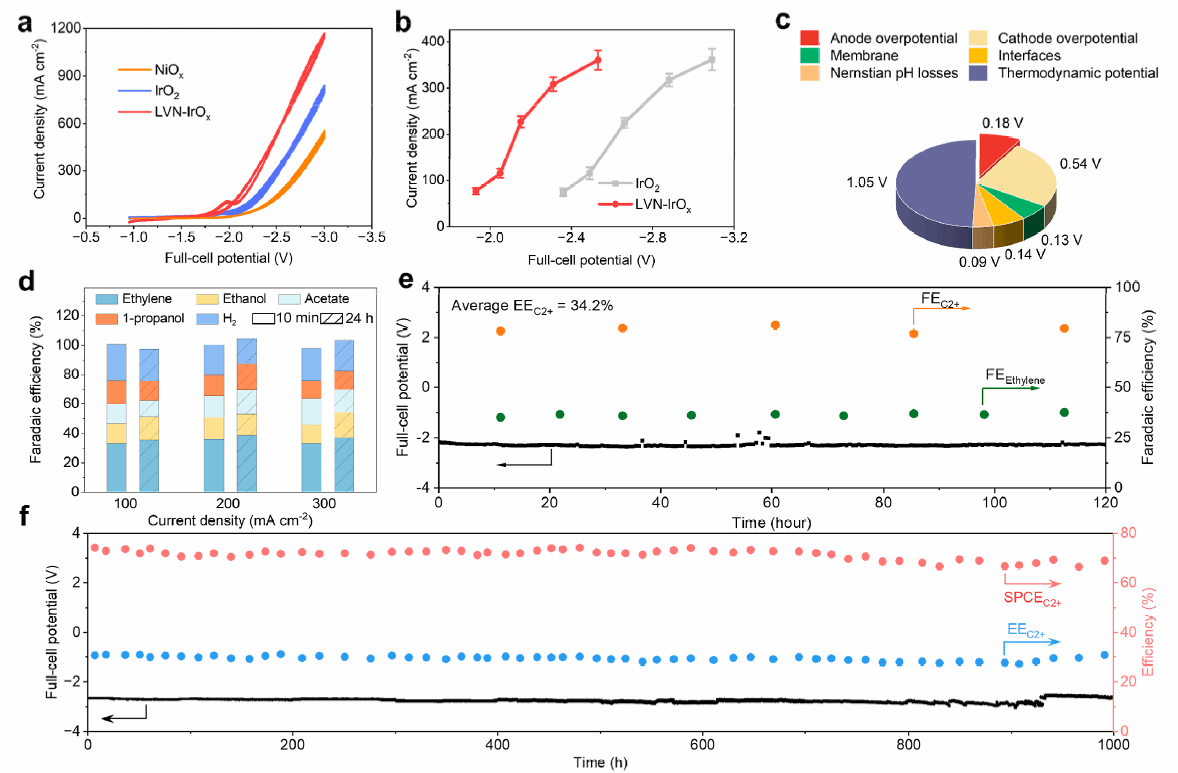
图5 (a) 不同阳极催化剂在MEA-CORR体系中的性能对比。(b) IrO₂与LVN-IrOx在不同电位下的电流密度响应。(c) MEA中, LVN-IrOx体系下的槽压组成分析。(d) LVN-IrOx体系在不同反应时间和电流密度下的产物分布。(e) LVN-IrOx作为阳极,商业Cu催化剂作为阴极在200 mA/cm2电流密度下的120 h稳定性测试。(f) LVN-IrOx作为阳极,自制多孔Cu粉作为阴极在1 A/cm2电流密度下的1000 h稳定性测试。
总而言之,实现高电流密度下具备能效高、碳利用率高且稳定性优异的CO电解过程,需同时优化阴极与阳极催化剂。我们系统研究了阳极催化剂的失活机制及其对CO还原性能的影响,进而开发出一种具备抗降解能力的低价态镍掺杂氧化铱阳极催化剂。将该稳定性优异的阳极催化剂与高比表面积的多孔铜阴极耦合应用,系统在1 A cm-2电流密度下实现了稳定运行超过1000小时,EEC2+达32%,SPCEC2+高达73%。该策略不仅有效降低了系统的资本和运行成本,同时突破了CO电解工业化过程中的关键瓶颈。研究结果突出表明,催化剂与系统的协同设计对推动高效、可持续CO电解技术的产业应用具有重要意义。




