

科学研究


可充电锂金属电池(LMBs)是下一代应用中最有前途的高能量密度电池技术之一。然而,不稳定的锂金属负极和枝晶形成会引发快速的容量衰减和安全风险,阻碍了LMBs的应用。构建LiF-rich SEI已被普遍认为是实现均匀锂沉积/剥离和抑制锂枝晶生长的关键。由于其高杨氏模量、高表面能和丰富的锂离子通量,LiF-rich SEI可以有效地分散局部电流,以均匀且低体积的方式调节锂成核,减少苔藓状枝晶生长的趋势。创建LiF-rich SEI的努力主要集中在提高电解液中的F含量。使用高浓度电解液(HCE)和局部高浓度电解液(LHCE)可以原位形成富含LiF的SEI,但其高成本极大地限制了其可行性。相反地,在RCEs中引入含氟添加剂(< 10 vol. %)(记为F-additive)似乎是一个更好的方法。
理想的F-additive,应符合如下三大指标,即高F/C摩尔比(更有可能增强锂金属界面处LiF的形成);兼容RCEs(溶解于电解液中而不发生分离);低LUMO(实现完全还原和在最初的几个循环中快速成膜)。然而,目前的主要瓶颈是当前的F-additive难以同时满足上述三个指标。例如,FEC是一种良好的SEI成膜添加剂,但其F/C摩尔比(0.33:1)较低。氟化醚溶剂,如HFE、TFETFE等,虽然具有高F/C摩尔比,但在电化学上对Li+配位和F分解表现为惰性,因此它们迄今为止只被开发为稀释剂。鉴于配位溶剂中轨道能级的变化,具有高F/C摩尔比的全氟取代的酸酐可以被视为理想的F-additive候选。Lucht等人在早期报道中证明,氟化酸酐添加剂能够参与SEI的形成,但SEI中的LiF含量并未如预期那样提高[J. Electrochem. Soc. 167, 110506 (2020)]。这种现象主要是由于氟化酸酐的高化学稳定性。因此,调整全氟取代酸酐的C−F键使其更容易断裂和还原,是开发F-additive的关键,但相关研究很少被报道。
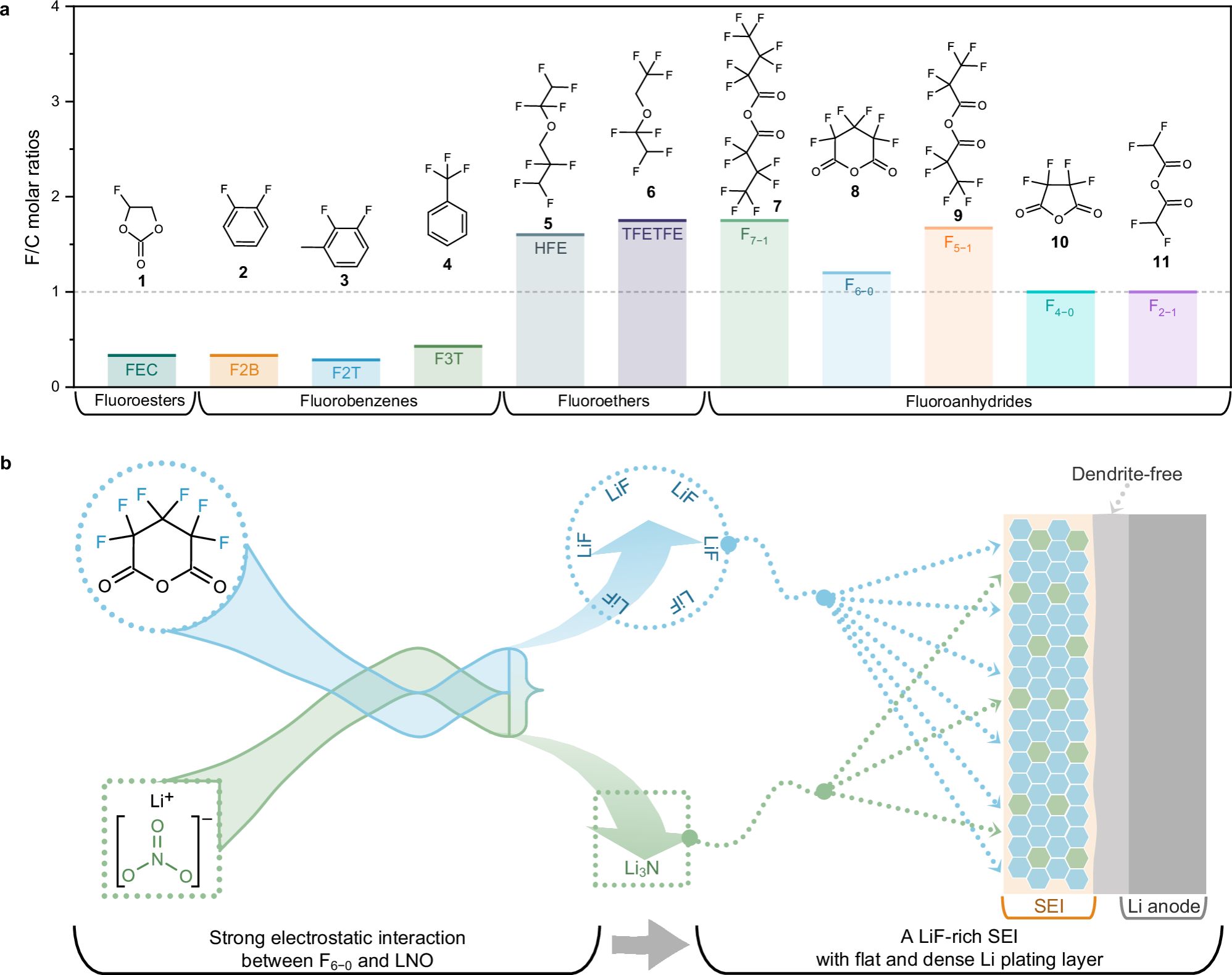
图1. 选定的F-additive工作机制及其与LNO的协同效应。
在此,上海交通大学变革性分子前沿科学中心梁正课题组研究了一系列具有不同F/C摩尔比的氟化添加剂,并证明了六氟戊二酸酐(F6−0)在常规碳酸酯电解液(RCEs)中形成LiF-rich SEI方面具有最佳潜能。为了改善F6−0的分解动力学,进一步在体系中引入了硝酸锂(LNO)作为辅助剂。结果表明,在F6−0/LNO协同作用下,F6−0的还原效率提高到91%,使得在仅添加4 vol. % F6−0/LNO (F6L)的RCE中,LMA形成了均匀的LiF-rich SEI。采用F6L的LiNi0.8Co0.1Mn0.1O2||Li(2.8~4.5 V)和LiNi0.5Mn1.5O4||Li(3.5~4.95 V)全电池也显示出比其他含氟添加剂更好的循环和倍率性能。
相关研究成果以“Screening of F-containing electrolyte additives and clarifying their decomposition routes for stable Li metal anodes”为题发表在Nature Communications上。第一作者为上海交通大学变革性分子前沿科学中心在读博士刘纪江、助理研究员郝伟、博士后方明明及访问学生Xin Chen。通讯作者为上海交通大学变革性分子前沿科学中心副教授梁正、助理研究员岳昕阳。唯一通讯单位为上海交通大学变革性分子前沿科学中心。
研究亮点1:设计了全新的F-additive:基于六氟戊二酸酐(F6−0)/硝酸锂(LNO)(记为F6L)的电解液添加剂,构建了作用于锂金属负极的LiF-rich SEI。
研究亮点2:破译了F6−0分解路径:基于DFT和in-situ FTIR分析,阐明了F6−0分解形成LiF过程中的机理。
研究亮点3:提出了F6−0还原率的定量测定方法:基于电荷守恒定律,通过积分还原过程的LSV曲线,精准测定了F6−0的还原率(从原来的3.1%提高到91%)。
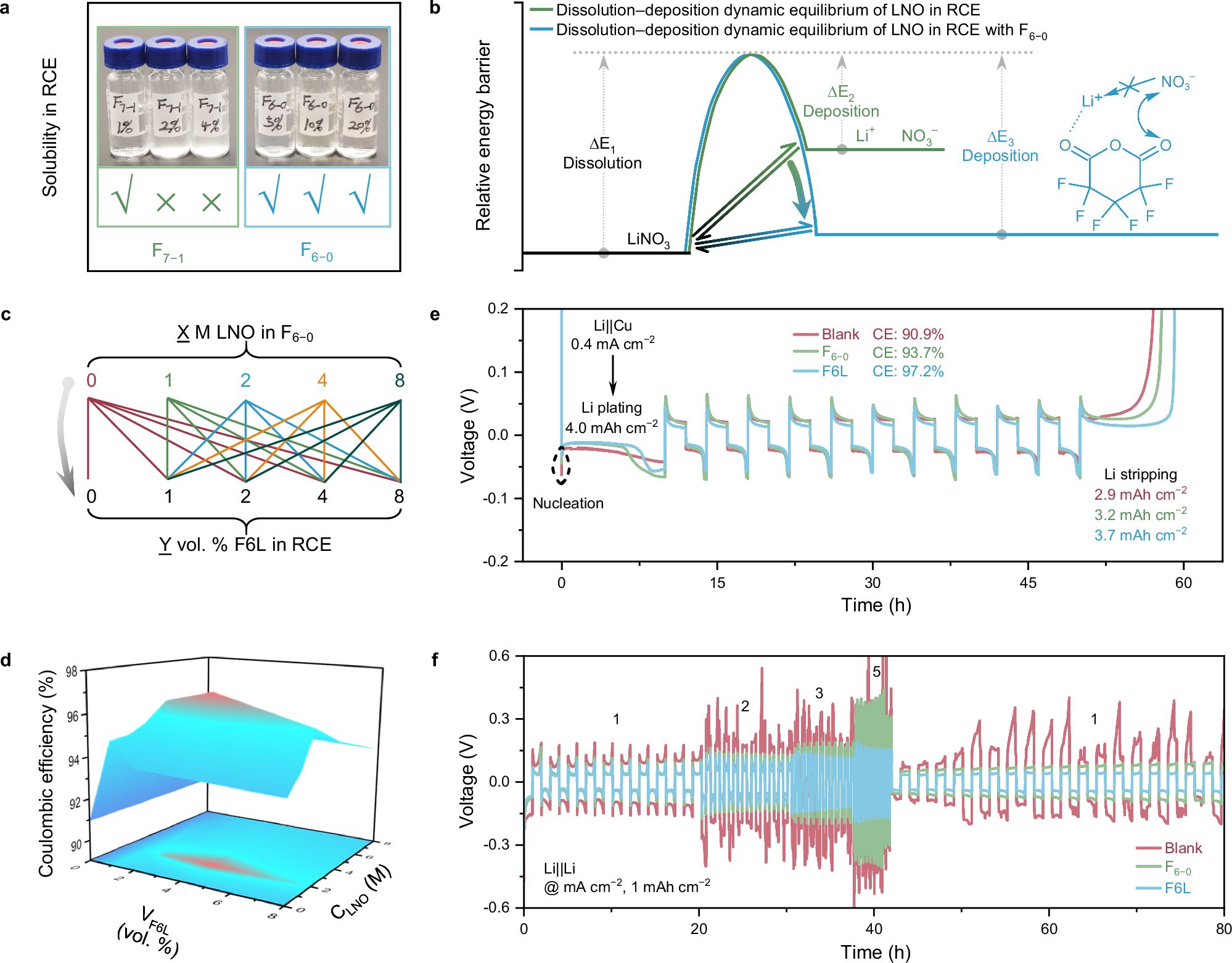
图2. 优化F6L的组成。
F6L成分优化:作者设计了21种不同比例的F6−0和LNO,以库仑效率(CE)为指标(参照Aurbach的方法计算)进行筛选,以获得F6L的最佳配方。最终,CE结果显示,在向RCE中添加含有4 vol. % F6L时,获得了最高的CE(97.2%),比空白RCE(Blank)高出6.3%,表明这种优化的F6L衍生的SEI在抑制锂枝晶生长方面的有效性。相比之下,仅添加4 vol. % F6−0或者LNO添加剂的电池的CE仅为93.7%和91.3%。优化的F6L、F6−0、RCE+LNO和Blank电池的锂成核极化电位分别在32.5、43.6、50.7和62.5 mV,这意味着在优化的F6L中形成的SEI可以促进锂成核。此外,所形成的SEI还具有高的锂离子传输动力学,这一点通过锂对称电池的倍率性能得到了证明。基于以上分析,选择4 vol. % F6L作为RCE中研究的F-additive。
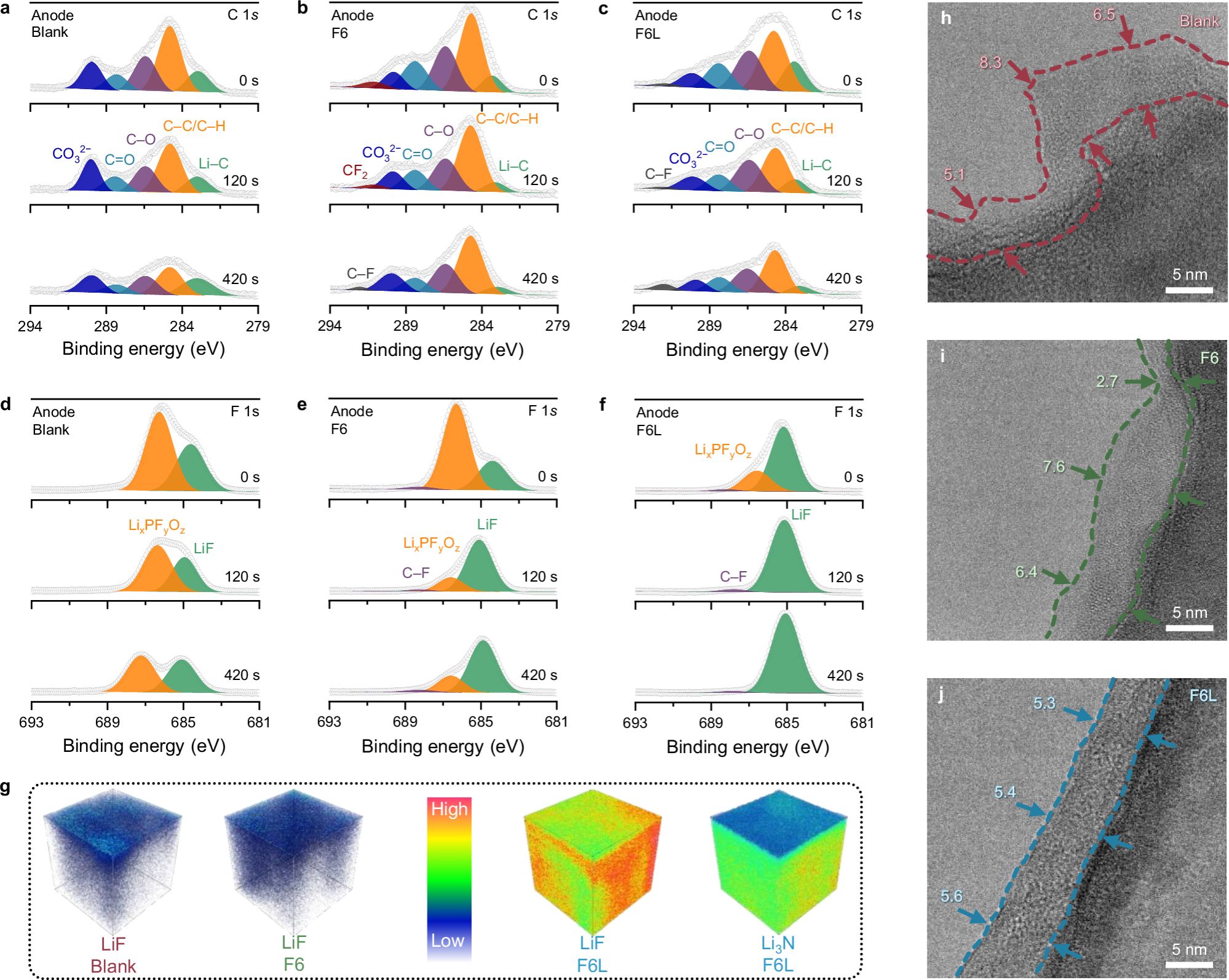
图3. 锂金属表面SEI表征。
F6L衍生SEI分析:C 1s XPS分析显示,相比于Blank,使用F6后,出现了一个新的峰(291.1 eV),可以归属于F6−0的F−C−F。当检测深度达到约5 nm时,F−C−F峰消失,C−F出现(292.0 eV),对应于F−C−F键的断裂。对于F6L样本,在所有厚度上,其C 1s XPS谱图显示F6−0的信号主要由C−F峰而非F−C−F峰反映,表明F6L的分解程度高于F6。在Blank的F 1s XPS谱图中,LiF和LixPFyOz源自RCE中LiPF6的分解,作为SEI的无机组分。使用F6可以在一定程度上增加SEI中LiF的含量,但F6的还原仍然很低,这一点通过更强的LixPFyOz峰值得到证实。相比之下,由于F6L的分解得到改善,其F 1s XPS谱图中的LiF峰在不同厚度下都占据主导地位。F6L XPS谱图中LixPFyOz峰值的降低意味着,由F6L优先分解形成的LiF层阻碍了LiPF6的还原。TOF-SIMS测试表明,在Blank中形成的SEI显示出LiF分布不均匀,这是由于LiPF6较弱的无机膜形成性能;在F6中,由于F6−0分解的动力学缓慢,SEI内LiF的分布也是不均匀的。而在F6L衍生的SEI中观察到一个三维均匀分布的LiF层,高下立判。这一结果与XPS分析吻合。cryo-TEM表征显示,在20圈循环后,无论是Blank(~6.5 nm)还是F6(~6.4 nm),SEI都呈现出粗糙的形态。相比之下,F6L衍生的SEI更薄(~5.5 nm)且均匀。简而言之,在LNO的辅助下,F6−0在RCE中分解成LiF的动力学加速,从而在锂金属负极上形成以LiF为主导的均匀且致密的SEI。
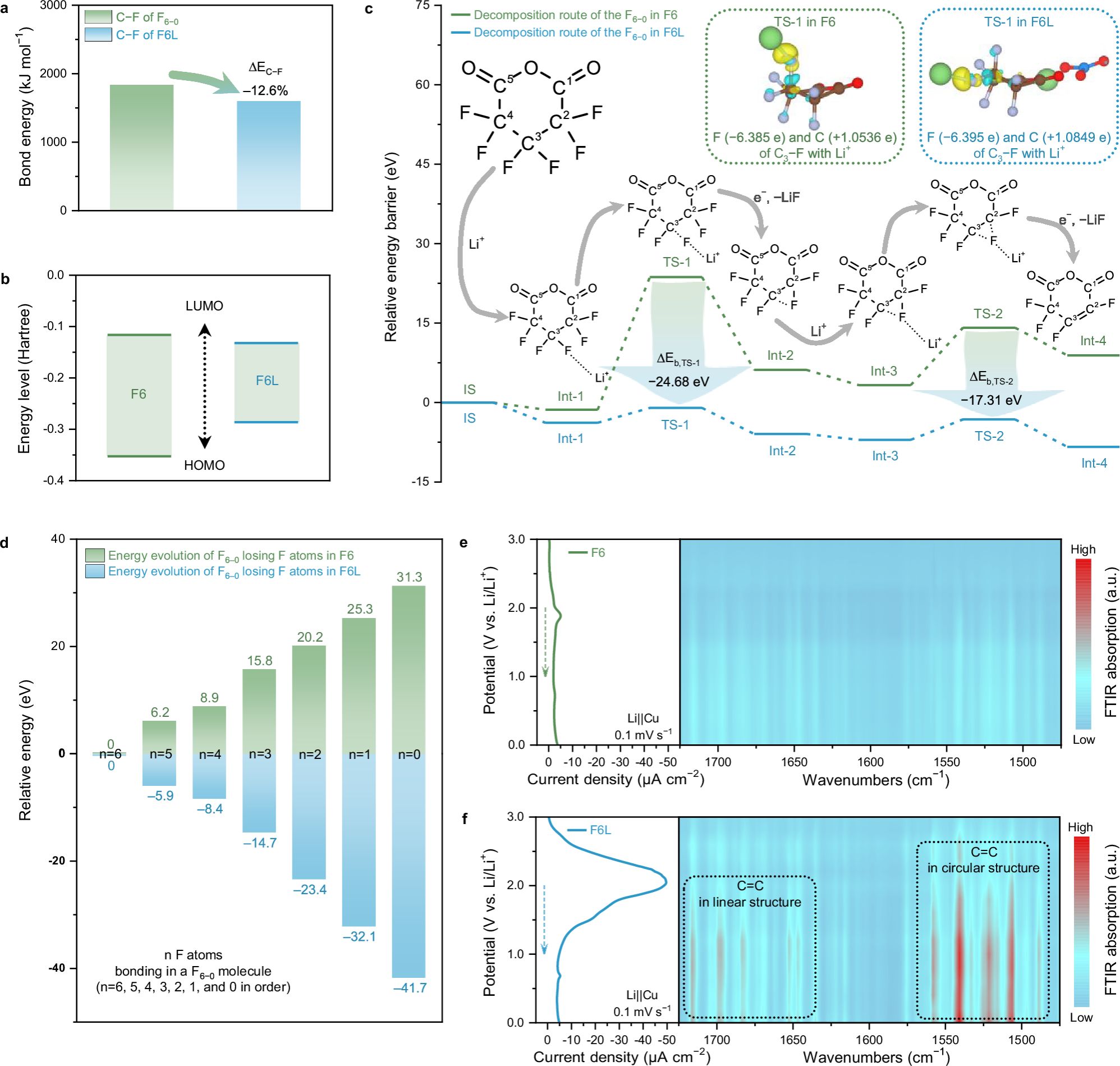
图4. 阐明F6−0的分解路径。
F6L分解化学:DFT计算表明,在静态条件下,当LNO分子接近F6−0分子时,F6−0中C−F的平均键能降低了12.6%,这表明F6−0/LNO之间较强的相互作用。F6−0的LUMO能级由于受到LNO的影响而降低,导致F6L中的F6−0比F6中的F6−0更倾向于在锂金属表面优先还原和分解。F6−0的分解过程可以划分为六个步骤,依次对应于LiF的形成。其中,前两个步骤是速率决定步骤(对应于分解过程中需要克服的最大能量障碍)。第一步是生成第一个LiF产物,涉及初始状态IS、中间态Int-1、过渡态TS-1和中间态Int-2。第二步包含了生成第二个LiF产物的过程,涉及中间态Int-3、过渡态TS-2和中间态Int-4。在第一步中,C3−F键在Li+的静电吸引下被削弱,然后电子丰富的F原子被捕获形成第一个LiF。这里主要的能量障碍,出现在从Int-1到TS-1的过程中(记作ΔEb, I1-T1)。F6和F6L的ΔEb, I1-T1分别为25.03和2.81 eV,这表明F6L中的第一个C3−F键比F6中的更容易断裂。F6−0中C3−F键的化学活性受到酸酐基团(O=C−O−C=O)引起的诱导效应的影响。C=O基团带来的诱导效应转移到C3原子上,并将C3−F键中的电子对吸引向C3原子,导致其比其他碳原子的正电荷状态相对较少。一旦F原子附近出现Li+,C−F键的电子密度将减少。因此,C3−F键的共价成分减少,离子成分增加,最终导致C3−F键断裂和LiF的形成。而对于F6L,NO3−在F6−0的C1=O基团附近的存在将π电子推向C1,减少了作用在C3上的诱导效应。因此,C3−F键中的电子对倾向于向F侧移动,增强了F原子的电子密度。在相同条件下,F6L中的C3−F键比F6−0中的C3−F键具有更高的离子成分,降低了F6L系统中的ΔEb, I1-T1。在第二步中,C2−F键中的F原子被Li+攻击并捕获,形成第二个LiF,同时产生的不饱和C2和C3形成了一个双键。与第一步类似,F6L中的NO3−加速了C2−F键的断裂。因此,F6L的ΔEb, I3-T2低至3.85 eV,而F6的ΔEb, I3-T2为10.83 eV。电化学in-situ FTIR支持了F6L中F6−0的高分解倾向。通过积分电化学还原LSV曲线并计算电荷平衡,确认F6L中F6−0的整体还原率从原来的3.1%提升到了91%。
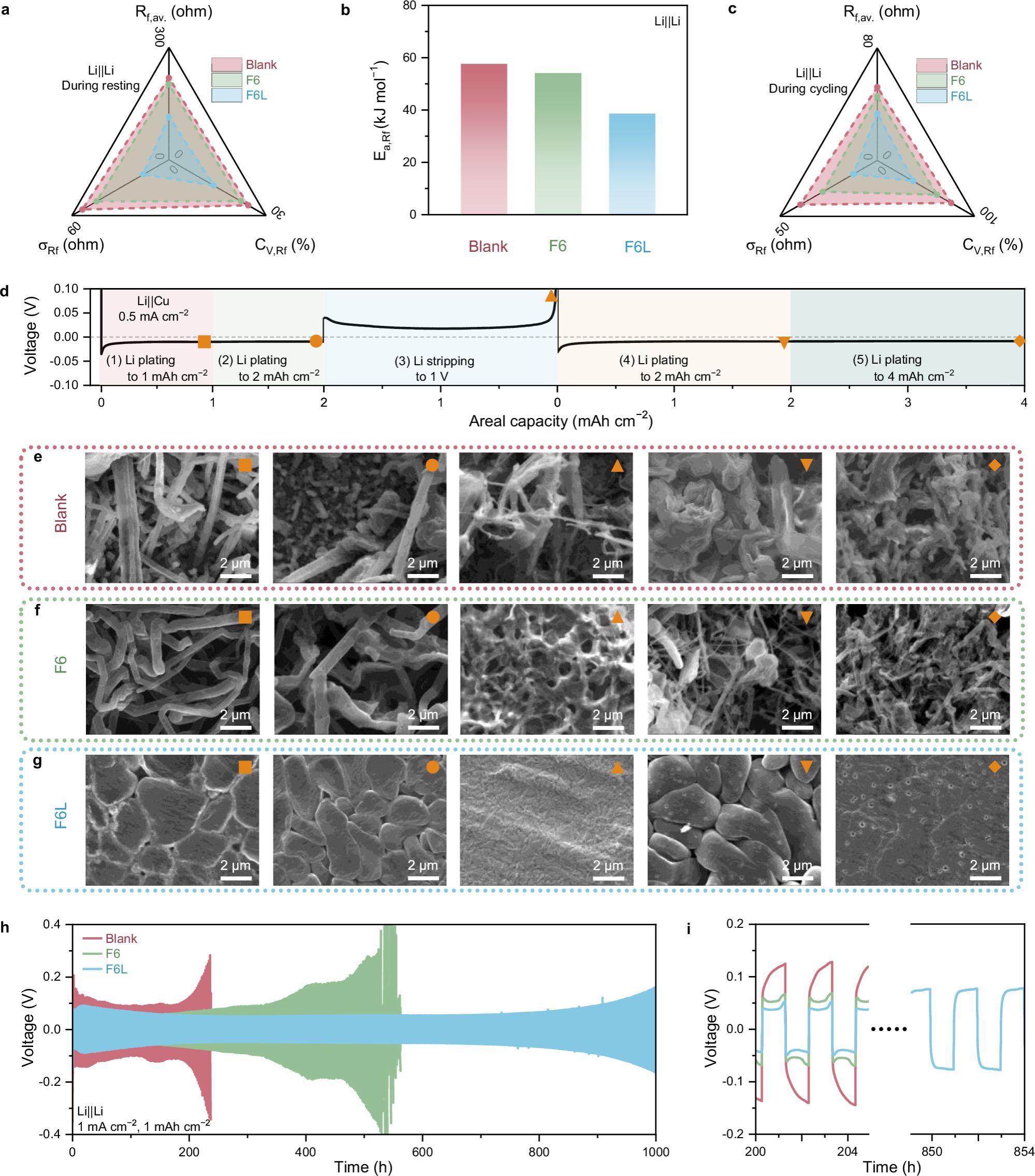
图5. F6L衍生SEI特性。
锂沉积/剥离分析:通过EIS记录了锂对称电池在静置期间界面电阻Rf的变化,以确定F6L衍生的LiF-rich SEI的热力学稳定性。通过统计学方法总结了锂穿过SEI的Rf数据,其中Rf,av.是平均Rf,σRf是标准差,CV,Rf是变异系数。σRf和CV,Rf反映了在静置期间Rf变化的幅度。F6L电池的σRf和CV,Rf低于Blank和F6电池,这表明F6L衍生的SEI具有优异的热力学稳定性。这是由于F6L的SEI中富含的LiF,在RCE中的溶解度比其他SEI的有机成分低,因此锂金属可以被有效地保护,从而免受电解液的侵蚀。与在Blank和F6中形成的SEI相比,F6L衍生的SEI以富含LiF及均匀结构为特征,具有快速的Li+传输能力,因此在F6L电池中观察到的Rf,av.较低。通过观察锂金属负极在沉积/剥离过程中Rf的变化,可以进一步评估SEI的动态结构稳定性。同样,从Blank、F6和F6L电池中得到的三组Rf数据计算出的三个统计学指标,即Rf,av.、σRf和CV,Rf,被用于横向对比。EIS谱图是在每一次锂沉积(0.25 mAh cm−2)或剥离(0.25 mAh cm−2)发生后测量的。显然,F6L电池的上述三个指标都低于其他两种电池,这表明F6L中的SEI不仅具有高结构稳定性以适应锂沉积和剥离的体积波动,而且还能限制有害的枝晶生长。在F6L中观察到的锂沉积形态与文献报道中关于LHCE或HCE的研究结果相似。在F6L中,经过1 mAh cm−2的锂沉积后,得到了相对平滑且具有典型岛状特点的形貌。在锂完全被剥离后,SEM显示了一个清晰而裸露的铜基底,表明了F6L中锂的高可逆循环性。锂对称电池实现了1000小时的稳定循环,且保持了较低的电压迟滞,这充分验证了F6L带来的性能提升。相比之下,F6和Blank电池的寿命仅为400小时和200小时。
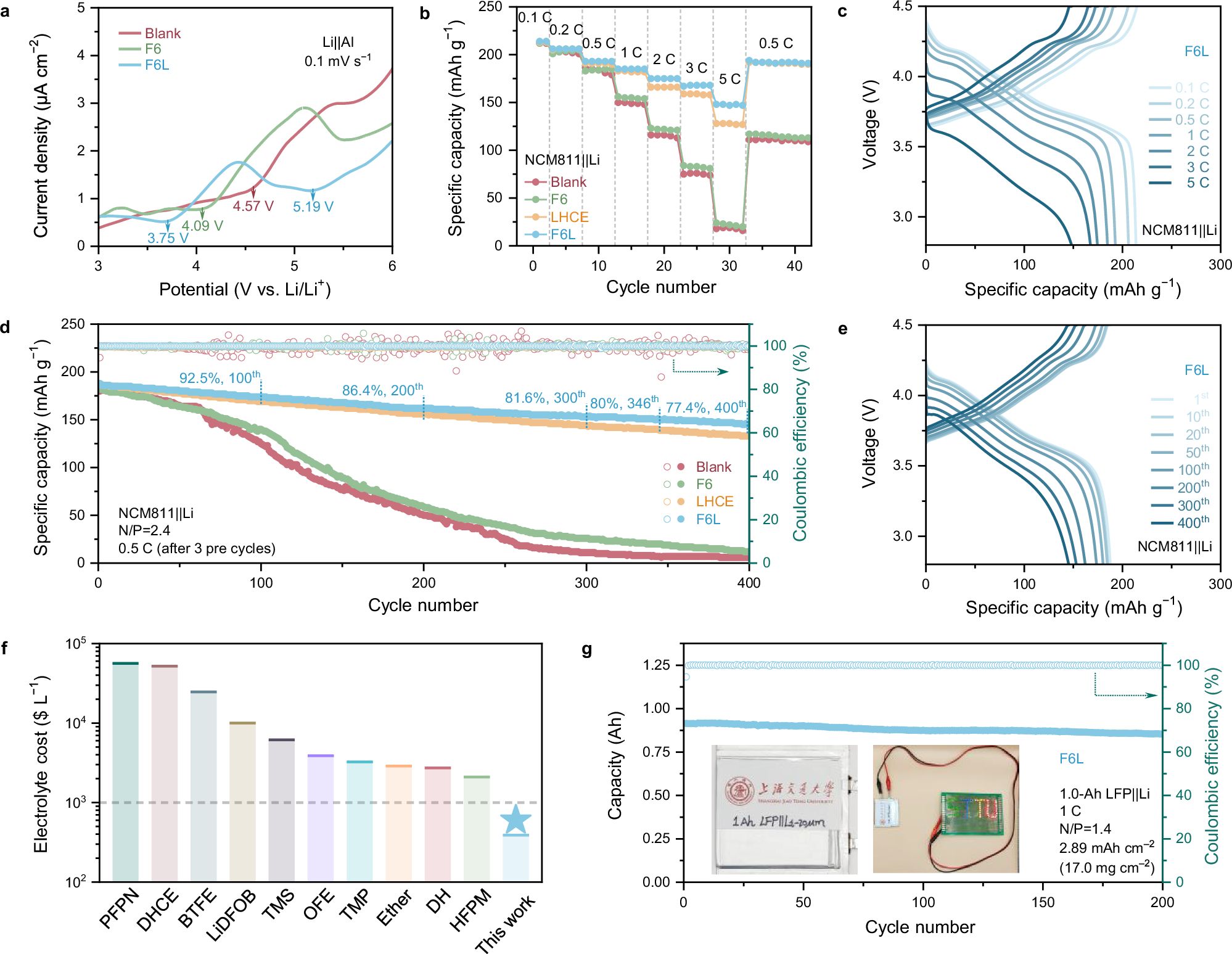
图6. 全电池性能。
电化学性能:凭借富含LiF的SEI的快速Li+传输,使用F6L的全电池在测试中实现了最低的极化和最高的充电状态,尤其是在2 C之后。尽管LHCE也可以获得LiF-rich SEI,但其较低的离子导率导致了5 C时电池容量的较大下降。长循环性能测试表明,使用F6L的NCM811||Li全电池能够稳定循环超过400圈,展现出188 mAh g−1的可逆容量、较低的极化和77.4%的容量保持率。与已经报道的锂金属电池电解液添加剂的研究相比,F6L在提高锂金属电池的倍率性能和使用寿命方面展现出优异的竞争力。同时,作者也评估了F6L在高电压LiNi0.5Mn1.5O4||Li全电池中的电化学性能,结果证实F6L在高电压锂金属电池中依然具有优异的稳定性。此外,作者还使用了自制的1.0-Ah LFP||Li软包电池,以评估F6L在实际应用中的可行性。F6L软包电池在1 C下经过200圈循环后,容量保持率达到了93.3%。
总结展望:在本研究中,通过在RCE中引入低成本的F6−0/LNO添加剂,来形成高质量的LiF-rich SEI,以均匀且无枝晶的方式调节锂的成核及沉积。研究结果表明,由C=O基团带来的对于C−F键的诱导效应,可以通过NO3−来降低,进而使得F6L中C−F键的断裂能垒低于单独的F6−0。这种协同效应有助于将F6−0在F6L中的整体还原比率从原始的3.1%提高到91%。因此,在RCE中添加4 vol. % F6L,能够有效地为锂金属负极赋予LiF-rich SEI。使用F6L的锂对称电池,在1 mA cm−2/1 mAh cm−2的条件下实现了超过1000小时的稳定循环,且电压迟滞较低。与其他对照组相比,F6L使得NCM811||Li和LNMO||Li全电池在高倍率和长期循环下的容量保持率都得到了显著地提高。更重要的是,F6L策略在成本上也具有优势。本研究提出的具有高F/C摩尔比、低分解能垒及合理成本的F-additives概念,为可持续和高性能锂金属电池的发展提供了一条可行的路线,同时也适用于其他先进的金属离子电池。
文章信息:Liu, J., Hao, W., Fang, M. et al. Screening of F-containing electrolyte additives and clarifying their decomposition routes for stable Li metal anodes. Nat Commun 15, 9356 (2024). https://doi.org/10.1038/s41467-024-53807-z




