

科学研究


自由基酰化反应具有反应条件温和、官能团兼容性良好的特点,是制备酮类化合物的一条重要途径。利用过渡金属催化的这类转化,在过去的几十年间发展迅速(图1a)。它们通常是由酰基自由基对金属复合物的加成或者酰基金属物种捕获烷基自由基来实现。此外,各种类型的酰基自由基前体与Giese受体之间在热化学或者光化学的引发下发生Giese型自由基酰化反应也得到了很好的研究(图1b)。然而,令人惊讶的是,涉及使用自由基酰化试剂的报道很少,而且通常反应效率不高(图1c)。在这一过程中,烷基自由基对酰化试剂加成会形成高能的氧中心自由基,这在热力学上是十分不利的。2021年,李超忠课题组报道了酰基膦酸酯可以作为一种高效的酰化试剂来捕获烯烃通过MHAT所产生的烷基自由基。然而,该试剂需要多步合成,并且尚未显示出对其他自由基前体的适用性。另外,磺酰肟醚也可用于间接自由基酰化,但需要额外的水解步骤才能得到酰化产物。为了避免高能氧中心自由基的产生,另一种策略是自由基-自由基偶联。这是一个快速发展的领域,其关键核心是将瞬态的酰基自由基转化为持久自由基,从而使捕获瞬态烷基自由基成为可能。氮杂环卡宾催化被证明是一种有效的途径(图1d)。Nagao和Ohmiya于2019年首次报道了利用氮杂环卡宾稳定酰基自由基以自由基-自由基偶联的方式实现酮类化合物的制备。此后,Studer等人在这一领域取得了较大进展。但是氮杂环卡宾催化剂通常也需要多步合成,其中一些步骤成本较高。因此,作者决定尝试使用一些苯甲酰化合物(如苯甲酰氰),作者认为其离去基团也可能稳定自由基,从而在没有任何氮杂环卡宾催化剂的帮助下实现预期的转化。
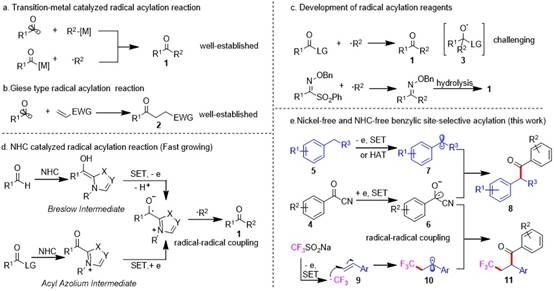
图1. 自由基酰化反应的最新进展和作者的假设
苄位C-H键存在于许多生物活性天然产物和畅销药物中,因此这种C-H键的直接官能团化在药物化学中有广泛用途。目前已经开发了多种策略来实现苄位C-H官能团化,包括酰化反应。目前已有报道使用光氧化还原/镍协同催化实现苄位C-H自由基酰化,作者希望开发一种不使用过渡金属镍的策略。作者还注意到Studer课题组最近利用光氧化还原/氮杂环卡宾双催化实现了这种转化。然而,他们的底物范围仅限于富电子芳烃。因此,作者决定开发一种以苯甲酰氰为自由基酰化试剂在无过渡金属镍和无氮杂环卡宾催化下实现苄位C-H的直接自由基酰化,该方法可广泛应用于富电子和缺电子芳烃。为了展示苯甲酰腈作为自由基酰化试剂的通用性,作者进一步将其应用于和苯乙烯的三组份三氟甲基化酰化反应中(图1e)。值得指出的是,作者的反应设计面临着两个挑战。首先,根据相关文献报道苯甲酰氰被还原容易形成自由基-自由基自偶联产物8aa-1,这是与作者期望的交叉偶联反应的竞争反应。其次,虽然苯甲酰氰商业易得,且价格便宜。但其他具有不同官能团取代的苯甲酰氰需要实验室制备。由于绝大多数苯甲酰氰对水敏感,因此在制备过程中存在麻烦。
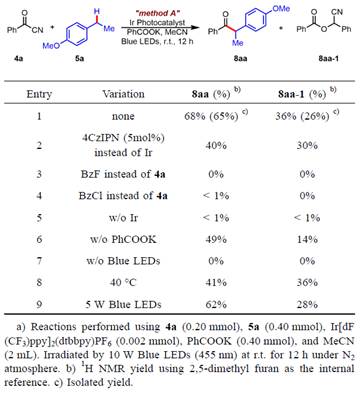
表1. 苯甲酰腈与富电子芳烃5a的条件筛选
作者选择使用苯甲酰腈与4-乙基苯甲醚作为模板底物进行了反应的尝试。具体实验中:以Ir[dF(CF3)ppy]2(dtbbpy)PF6作为光敏剂,苯甲酸钾作为碱,乙腈作为溶剂在蓝光的激发下,以65%的分离产率获得目标化合物,并且以26%的产率分离出了苯甲酰腈自偶联副产物8aa-1(Method A)。
随后,作者考察了苯甲酰腈与富电子芳烃反应的底物范围(图2)。总体而言,该反应具有较好的官能团兼容性,同时具有优异的区域选择性。最后,为了进一步证明作者方法的适用性,作者成功将这种自由基-自由基交叉偶联方法应用于多巴胺、雌马酚、山道年等生物活性分子苄位C-H键的后期酰化修饰。
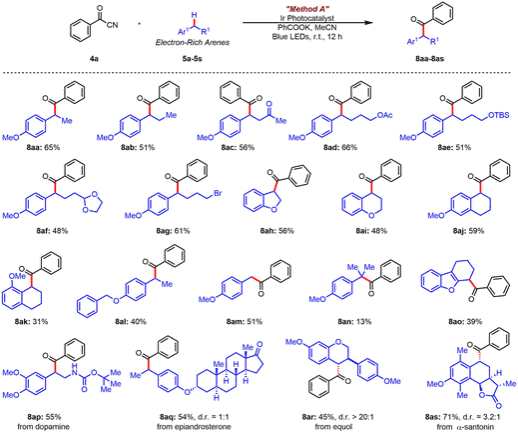
图2. 苯甲酰腈与富电子芳烃的底物拓展
令人失望的是,当芳烃为电中性或者缺电子时,芳香环中的电子密度降低使得难以被激发的光催化剂氧化,因此只能获得痕量的目标产物。由此作者另辟蹊径,以溴化钠作为氢原子转移(HAT)试剂,通过HAT策略成功解决了苯甲酰腈与电中性或者缺电子芳烃之间的偶联难题(Method B)。接下来,作者考察了苯甲酰腈与电中性或者缺电子芳烃之间的底物范围(图3)。总体而言,反应的官能团兼容性良好,但是与富电子芳烃不同,作者并没有观测很好的区域选择性,同时作者发现当反应底物含有相同化学环境的苄位C-H时,反应只能得到单酰化产物。对于反应条件稍作调整,也可用于实现烯丙位C-H键的酰化反应。
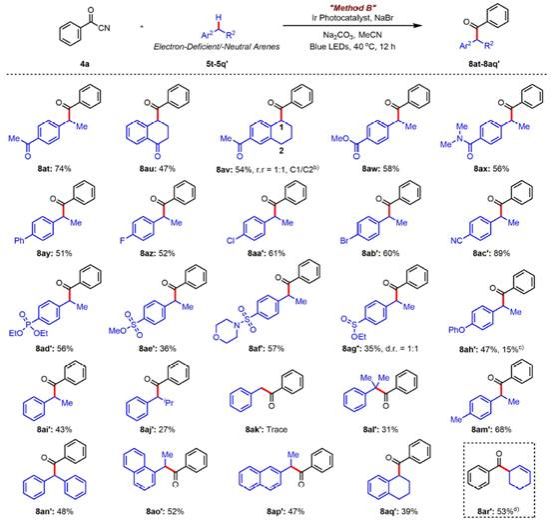
图3. 苯甲酰腈与电中性或者缺电子芳烃的底物拓展
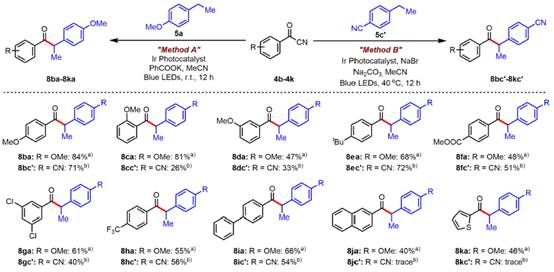
图4. 苯甲酰腈的官能团兼容性研究
紧接着,作者分别在Method A和Method B的条件下考察了苯甲酰腈的官能团兼容性(图4)。整体而言,在Method A的条件下,无论带有富电子基团还是缺电子基团的苯甲酰腈都能高效地转化。而在Method B的条件下,个别底物会发生快速地苯甲酰腈自偶联,从而导致交叉偶联的效率下降或者不能参与反应。
在发展了苄位C-H键直接自由基酰化的基础上,为了进一步展示苯甲酰腈作为新型自由基酰化试剂的通用性,作者决定尝试更具挑战性的苯乙烯三组份三氟甲基化酰化反应。虽然,通过氮杂环卡宾催化已经实现了该类反应,但无氮杂环卡宾催化,甚至无需额外加碱的方法仍未报道。幸运的是,作者使用苯甲酰腈,苯乙烯和三氟甲基亚磺酸钠为底物,Ir(ppy)2(dtbbpy)PF6为光敏剂,丙酮和N,N-二甲基甲酰胺作为混合溶剂,在蓝光照射下,以55%的收率获得对应的苯乙烯酰三氟甲基化产物(Method C)。
随后,作者对这一类型的反应也进行了底物的探究(图5)。作者发现即使是α位有取代的苯乙烯也能很好的发生反应,这是Studer课题组所发展的方法没有做到的。富电子的苯乙烯更有利于亲电性三氟甲基自由基的加成,因此显示出了更好的转化效率。以1,1-二苯乙烯作为自由基捕获试剂时,无论苯甲酰腈上取代基的电性如何,都能很好地得到三组分偶联的产物。
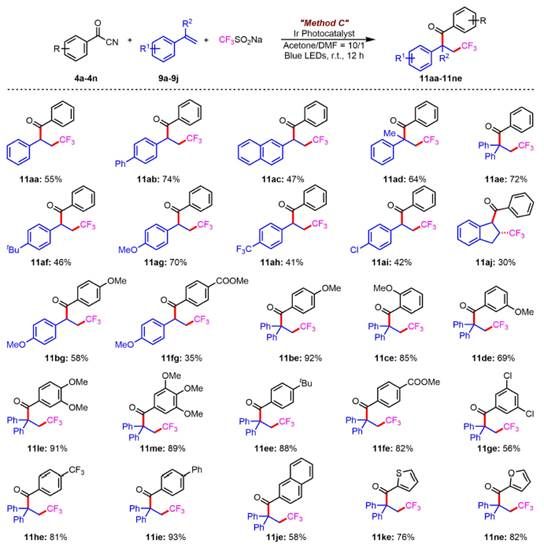
图5. 苯乙烯自由基酰三氟甲基化研究
最后,作者对反应的机理进行了研究(图6)。作者分别在Method A和Method B的条件下加入TEMPO作为自由基抑制剂,目标反应被抑制,通过HRMS作者检测到了苄基自由基被TEMPO捕获的产物,同时作者也分离出了苯甲酰基被TEMPO捕获的产物,由此作者认为反应的历程中存在苯甲酰基自由基以及苄基自由基(图6a)。随后,作者研究了自由基钟实验,成功分离得到关环产物,再次验证了苄基自由基的存在(图6b)。通过阴离子实验,虽然作者不能完全排除反应的历程中存在离子型酰化的过程,但是自由基型酰化反应在该条件下应为主导过程。
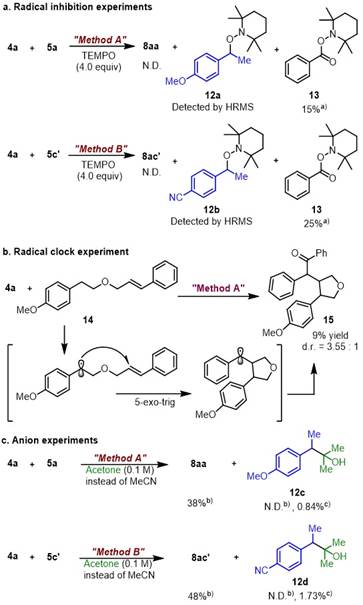
图6. 机理研究
基于上述的结果以及文献的报道,通过EPR等实验验证,作者提出了如下可能的机理(图7)。总结来看,苯甲酰腈被光敏剂还原生成寿命较长的关键中间体III,由此来捕获通过光直接氧化芳环、HAT攫取苄位C-H或者苯乙烯被三氟甲基自由基加成而产生的瞬态苄位自由基,通过自由基-自由基偶联的方式实现相应的酮的构建。
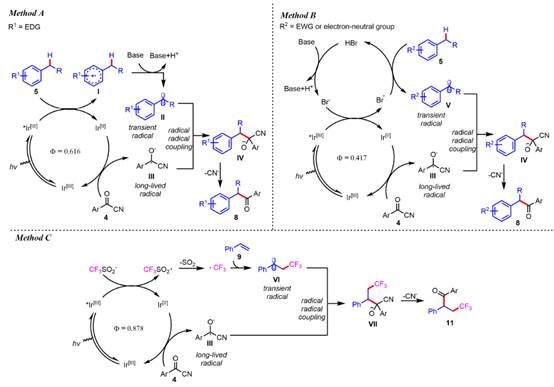
图7. 可能的机理
综上所述,在理性设计的基础上,作者首次解锁了离子型酰化试剂苯甲酰氰的自由基酰化新功能。通过SET和HAT策略,作者开发了苯甲酰氰参与的两种不同的可见光光氧化还原催化的苄位C-H键直接酰化反应。为了进一步证明该试剂的通用性,苯甲酰氰被成功应用于更具挑战性的苯乙烯三组份酰三氟甲基化反应。反应在温和、无过渡金属和无氮杂环卡宾的参与的情况下进行,具有高官能团耐受性。此外,这两种苄位C-H键酰化策略都具有广泛的底物范围,其中不同的富电子、电中性和缺电子芳烃都可以兼容。通过这种方法,可以实现多个天然产物衍生物的苄位C-H键直接酰化反应。值得注意的是,分子中的氰基不仅作为离去基,而且作为一种稳定自由基阴离子中间体的官能团,这使得它相当于一个长寿的自由基。进一步将这种自由基酰化试剂,应用于其他类型自由基酰化反应的尝试目前正在作者的实验室开展。
该研究成果近期以“Benzoyl cyanide, a general radical acylating reagent for photoredox-catalyzed benzylic site-selective acylation reactions”为题,在《Science China Chemistry》上线发表(DOI:10.1007/s11426-024-2309-5)。上海交通大学博士研究生朱兆栋为论文第一作者,上海交通大学吴晶晶副教授为论文的通讯作者。作者特别感谢英国布里斯托大学的Adam Noble教授和Kay Yeung博士对本文的有益的讨论和校对。作者感谢苏州大学的鲍晓光教授、吴新鑫教授和博士生刘田田对相关DFT计算部分提供支持。作者感谢上海交通大学张兆国教授分享实验室。




